Diagonalisasi kuantum berbasis sampel untuk Hamiltonian kimia
Estimasi penggunaan: kurang dari satu menit pada prosesor Heron r2 (CATATAN: Ini hanya estimasi. Waktu aktual bisa berbeda.)
Hasil pembelajaran
Setelah menyelesaikan tutorial ini, pengguna diharapkan memahami:
- Cara menggunakan SQD Qiskit addon untuk mengaproksimasi energi ground state sistem molekuler menggunakan bitstring yang disampling dari unit pemrosesan kuantum (QPU).
- Cara menggunakan ffsim untuk membangun Circuit local unitary cluster Jastrow (LUCJ) untuk simulasi kimia kuantum.
Prasyarat
Kami merekomendasikan pengguna untuk memahami topik-topik berikut sebelum menjalani tutorial ini:
- Kimia kuantum dan kuantisasi kedua
- Menggunakan Sampler primitive untuk mengambil sampel dari Circuit kuantum
Latar Belakang
Dalam tutorial ini, kita akan menunjukkan cara memproses sampel kuantum yang berisik untuk mengaproksimasi ground state molekul nitrogen pada panjang ikatan kesetimbangan, dengan menggunakan SQD Qiskit addon untuk mengimplementasikan algoritma diagonalisasi kuantum berbasis sampel (SQD). Detail lebih lanjut tentang perangkat lunaknya dapat ditemukan di dokumentasi yang sesuai, termasuk contoh sederhana untuk memulai.
Tutorial ini direkomendasikan bagi pengguna yang sudah familiar dengan kimia kuantum: khususnya, keakraban dalam menemukan energi ground state suatu molekul. Untuk penjelasan mendetail tentang alur kerja, lihat kursus algoritma diagonalisasi kuantum.
SQD adalah teknik untuk menemukan nilai eigen dan vektor eigen dari operator kuantum, seperti Hamiltonian sistem kuantum, dengan menggunakan komputasi kuantum dan komputasi klasik terdistribusi bersama-sama. Komputasi klasik terdistribusi digunakan untuk memproses sampel yang diperoleh dari prosesor kuantum, serta untuk memproyeksikan dan mendiagonalisasi Hamiltonian target dalam subruang yang dibentangkan oleh sampel tersebut. Alur kerja berbasis SQD memiliki langkah-langkah berikut:
- Pilih ansatz sirkuit dan terapkan pada komputer kuantum ke sebuah state referensi (dalam kasus ini, state Hartree-Fock).
- Sampel bitstring dari state kuantum yang dihasilkan.
- Jalankan prosedur self-consistent configuration recovery pada bitstring untuk mendapatkan aproksimasi ke ground state.
SQD diketahui bekerja dengan baik ketika eigenstate target bersifat jarang: fungsi gelombang didukung dalam sekumpulan state basis yang ukurannya tidak bertambah secara eksponensial seiring dengan ukuran masalah.
Kimia kuantum
Hamiltonian sistem molekuler dapat ditulis sebagai
di mana dan adalah bilangan kompleks yang disebut integral molekuler yang dapat dihitung dari spesifikasi molekul menggunakan sebuah program komputer. Dalam tutorial ini, kita menghitung integral menggunakan paket perangkat lunak PySCF.
Untuk detail tentang cara Hamiltonian molekuler diturunkan, konsultasikan buku teks kimia kuantum (misalnya, Modern Quantum Chemistry oleh Szabo dan Ostlund). Untuk penjelasan tingkat tinggi tentang bagaimana masalah kimia kuantum dipetakan ke komputer kuantum, lihat kuliah Mapping Problems to qubits dari Qiskit Global Summer School 2024.
Ansatz local unitary cluster Jastrow (LUCJ)
SQD memerlukan ansatz sirkuit kuantum untuk mengambil sampel. Dalam tutorial ini, kita akan menggunakan ansatz local unitary cluster Jastrow (LUCJ) karena kombinasinya antara motivasi fisik dan keramahan terhadap perangkat keras. Kita akan menggunakan ffsim untuk membangun Circuit ansatz.
Ansatz LUCJ beradaptasi dengan QPU yang memiliki konektivitas qubit terbatas. Orbital-orbital spin dipetakan ke qubit sedemikian rupa sehingga ansatz tidak memerlukan routing dengan gate SWAP. Perangkat keras IBM® memiliki topologi qubit kisi heavy-hex, di mana kita dapat mengadopsi pola "zig-zag", yang digambarkan di bawah. Dalam pola ini, orbital dengan spin yang sama dipetakan ke qubit dengan topologi garis (lingkaran merah dan biru), dan koneksi antara orbital dengan spin berbeda hadir setiap 4 orbital spasial ke-4, dengan koneksi yang difasilitasi oleh qubit ancilla (lingkaran ungu).
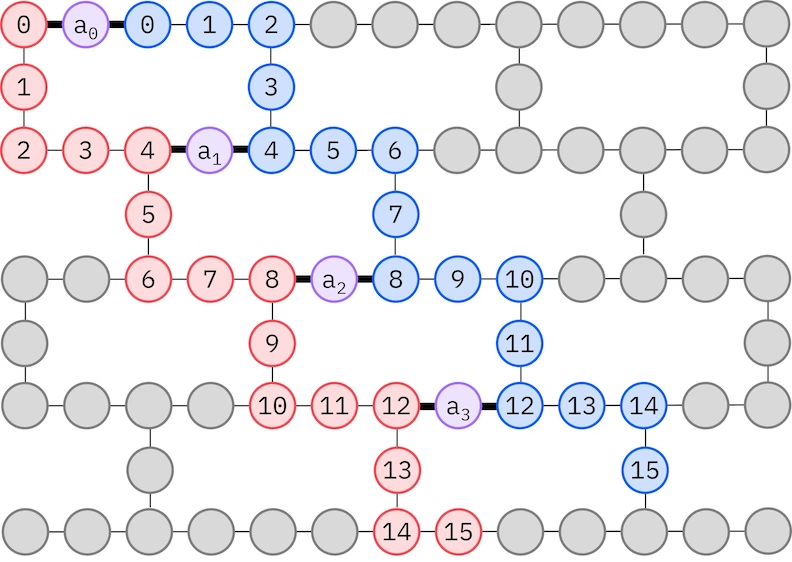
Self-consistent configuration recovery
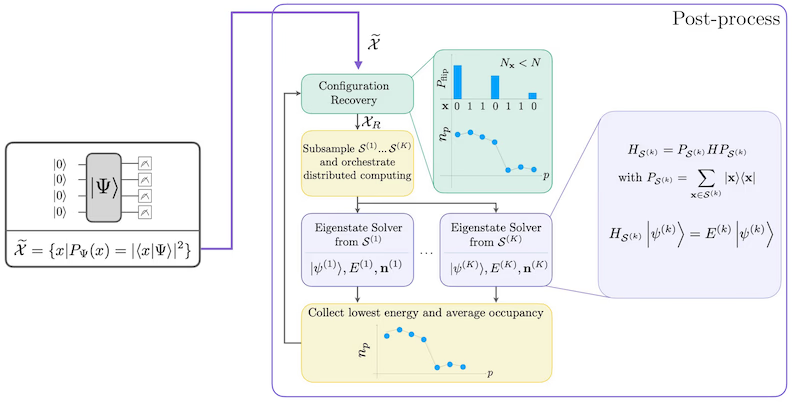
Prosedur self-consistent configuration recovery dirancang untuk mengekstrak sinyal sebanyak mungkin dari sampel kuantum yang berisik. Karena Hamiltonian molekuler mengkonservasi jumlah partikel dan spin Z, masuk akal untuk memilih ansatz sirkuit yang juga mengkonservasi simetri-simetri ini. Ketika diterapkan ke state Hartree-Fock, state yang dihasilkan memiliki jumlah partikel dan spin Z yang tetap dalam pengaturan tanpa noise. Oleh karena itu, bagian spin- dan spin- dari setiap bitstring yang disampling dari state ini seharusnya memiliki Hamming weight yang sama seperti pada state Hartree-Fock. Karena adanya noise pada prosesor kuantum saat ini, beberapa bitstring yang diukur akan melanggar sifat ini. Bentuk sederhana dari postseleksi akan membuang bitstring-bitstring ini, tetapi hal itu boros karena bitstring tersebut mungkin masih mengandung sinyal. Prosedur self-consistent recovery berupaya memulihkan sebagian sinyal tersebut dalam post-processing. Prosedur ini bersifat iteratif dan memerlukan estimasi awal rata-rata penghunian setiap orbital dalam ground state sebagai input, yang pertama kali dihitung dari sampel mentah. Prosedur ini dijalankan dalam sebuah loop, dan setiap iterasi memiliki langkah-langkah berikut:
- Untuk setiap bitstring yang melanggar simetri yang ditentukan, balik bit-nya dengan prosedur probabilistik yang dirancang untuk membawa bitstring lebih dekat ke estimasi saat ini dari rata-rata penghunian orbital, untuk mendapatkan bitstring baru.
- Kumpulkan semua bitstring lama dan baru yang memenuhi simetri, dan subsample subset dari ukuran tetap yang dipilih sebelumnya.
- Untuk setiap subset bitstring, proyeksikan Hamiltonian ke subruang yang dibentangkan oleh vektor basis yang sesuai (lihat bagian sebelumnya untuk deskripsi vektor basis ini), dan hitung estimasi ground state dari Hamiltonian yang diproyeksikan pada komputer klasik.
- Perbarui estimasi rata-rata penghunian orbital dengan estimasi ground state yang memiliki energi terendah.
Diagram alur kerja SQD
Alur kerja SQD digambarkan dalam diagram berikut:
Persyaratan
Sebelum memulai tutorial ini, pastikan kamu telah menginstal yang berikut:
- Qiskit SDK v1.0 atau lebih baru, dengan dukungan visualisasi
- Qiskit Runtime v0.22 atau lebih baru (
pip install qiskit-ibm-runtime) - SQD Qiskit addon v0.11 atau lebih baru (
pip install qiskit-addon-sqd) - ffsim v0.0.75 atau lebih baru (
pip install ffsim)
Pengaturan
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import math
import ffsim
import matplotlib.pyplot as plt
import numpy as np
import pyscf
import pyscf.cc
import pyscf.mcscf
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.primitives import StatevectorSampler
from qiskit.providers.fake_provider import GenericBackendV2
from qiskit_ibm_runtime import QiskitRuntimeService
from qiskit_ibm_runtime import SamplerV2 as Sampler
Contoh simulator skala kecil
Dalam tutorial ini, kita akan mencari aproksimasi terhadap ground state molekul nitrogen mendekati jarak ikatan kesetimbangannya. Kita pertama menggunakan basis set STO-6G yang kecil agar bisa mensimulasikan eksperimen dan memastikannya berfungsi.
Langkah 1: Petakan input klasik ke masalah kuantum
Pertama, kita tentukan molekul dan properti-propertinya.
# Specify molecule properties
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="sto-6g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Compute exact energy using FCI
reference_energy = cas.run().e_tot
print(f"norb = {norb}")
print(f"nelec = {nelec}")
converged SCF energy = -108.464957764796
CASCI E = -108.595987350986 E(CI) = -32.4115475088426 S^2 = 0.0000000
norb = 8
nelec = (5, 5)
Sebelum membangun Circuit ansatz LUCJ, kita terlebih dahulu melakukan perhitungan CCSD pada sel kode berikut. Amplitudo dan dari perhitungan ini akan digunakan untuk menginisialisasi parameter ansatz.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -108.5933309085008 E_corr = -0.1283731437052354
Sekarang, kita gunakan ffsim untuk membuat Circuit ansatz. Karena molekul kita memiliki state Hartree-Fock closed-shell, kita gunakan varian spin-balanced dari ansatz UCJ, yaitu UCJOpSpinBalanced. Kita set optimize=True pada metode from_t_amplitudes untuk mengaktifkan double-factorization "terkompresi" dari amplitudo (lihat The local unitary cluster Jastrow (LUCJ) ansatz di dokumentasi ffsim untuk detailnya).
Karena ansatz LUCJ beradaptasi dengan konektivitas yang tersedia pada QPU, kita harus menginisialisasi backend QPU sebelum membuat ansatz. Untuk saat ini, kita akan membuat backend generik dengan coupling map heavy-hex dan gate set yang secara alami diurai oleh ansatz LUCJ. Kemudian, kita akan menggunakan ffsim.qiskit.generate_lucj_pass_manager untuk membuat pass manager yang dikhususkan untuk melakukan transpile ansatz LUCJ ke backend yang diberikan sesuai dengan layout "zig-zag" yang dijelaskan di bagian latar belakang tentang ansatz LUCJ. Fungsi ini menggunakan heuristik penilaian untuk meminimalkan error yang terkait dengan layout yang dipilih, yang penting jika backend kamu adalah QPU nyata atau simulator dengan model noise. Selain mengembalikan pass manager, fungsi ini juga mengembalikan pasangan interaksi alpha-beta yang dapat diimplementasikan pada hardware. Jika tidak semua pasangan dapat diimplementasikan, fungsi ini mengeluarkan peringatan.
import warnings
from qiskit.transpiler import CouplingMap
warnings.formatwarning = lambda msg, *args, **kwargs: f"Warning: {msg}\n"
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
coupling_map = CouplingMap.from_heavy_hex(3)
backend = GenericBackendV2(
coupling_map.size(),
coupling_map=coupling_map,
basis_gates=["cp", "xx_plus_yy", "p", "x", "swap"],
)
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
Langkah 2: Optimalkan untuk eksekusi hardware kuantum
Selanjutnya, kita optimalkan Circuit untuk hardware target. Biasanya, langkah ini melibatkan inisialisasi backend hardware dan pass manager untuk backend tersebut. Namun, karena ansatz LUCJ sudah disesuaikan dengan konektivitas hardware, kita sudah melakukan tindakan-tindakan tersebut di langkah sebelumnya. Yang perlu dilakukan hanyalah menjalankan pass manager pada Circuit untuk melakukan transpile menjadi ISA circuit yang dapat langsung dieksekusi di QPU.
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
Gate counts: OrderedDict({'xx_plus_yy': 86, 'p': 16, 'measure': 16, 'cp': 15, 'x': 10, 'swap': 2, 'barrier': 1})
Langkah 3: Eksekusi menggunakan Qiskit primitives
Setelah mengoptimalkan Circuit untuk eksekusi hardware, kita siap menjalankannya di hardware target dan mengumpulkan sampel untuk estimasi energi ground state. Karena kita hanya memiliki satu sirkuit, kita akan menggunakan mode eksekusi Job dari Qiskit Runtime dan mengeksekusi Circuit kita.
rng = np.random.default_rng()
sampler = StatevectorSampler(seed=rng)
job = sampler.run([isa_circuit], shots=100_000)
Warning: Trying to add QuantumRegister to a QuantumCircuit having a layout
primitive_result = job.result()
pub_result = primitive_result[0]
Langkah 4: Pasca-proses dan kembalikan hasil dalam format klasik yang diinginkan
Metrik yang berguna untuk menilai kualitas output QPU adalah jumlah konfigurasi valid yang dikembalikan. Konfigurasi valid memiliki jumlah partikel dan spin Z yang benar, yang berarti separuh kanan dari bitstring memiliki Hamming weight yang sama dengan jumlah elektron spin-up, dan separuh kiri memiliki Hamming weight yang sama dengan jumlah elektron spin-down. Sel berikut menghitung fraksi konfigurasi yang disampling yang valid.
def is_valid_bitstring(
bitstring: str, norb: int, nelec: tuple[int, int]
) -> bool:
n_alpha, n_beta = nelec
return (
len(bitstring) == 2 * norb
and bitstring[norb:].count("1") == n_alpha
and bitstring[:norb].count("1") == n_beta
)
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
Fraction of sampled configurations that are valid: 1.0
Semua bitstring valid karena kita mengambil sampel Circuit pada simulator tanpa noise. Ketika menjalankan pada QPU yang berisik, fraksi tersebut akan kurang dari satu, tetapi diharapkan lebih besar dari fraksi yang akan didapat jika bitstring disampling secara acak seragam, yang dihitung pada sel berikut.
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
Expected fraction of valid configurations from uniformly random bitstrings: 0.0478515625
Sekarang, kita estimasi energi ground state dari Hamiltonian menggunakan fungsi diagonalize_fermionic_hamiltonian. Fungsi ini melakukan prosedur configuration recovery yang self-consistent untuk secara iteratif menyempurnakan sampel kuantum yang berisik guna meningkatkan estimasi energi. Kita berikan fungsi callback agar bisa menyimpan hasil antara untuk analisis lebih lanjut. Lihat dokumentasi API untuk penjelasan argumen-argumen diagonalize_fermionic_hamiltonian.
Di sini, kita menggunakan argumen initial_occupancies pada diagonalize_fermionic_hamiltonian untuk menentukan konfigurasi Hartree-Fock sebagai tebakan awal untuk penghunian orbital dalam ground state. Pendekatan ini masuk akal untuk sistem di mana ground state memiliki dukungan signifikan pada konfigurasi Hartree-Fock, tetapi mungkin tidak cocok dalam situasi lain, meskipun metode komputasi yang lebih canggih mungkin menghasilkan tebakan awal yang lebih baik dalam kasus-kasus tersebut. Menentukan initial_occupancies juga memungkinkan configuration recovery berjalan bahkan jika tidak ada konfigurasi valid yang disampling, seperti yang mungkin terjadi saat mengambil sampel dari Circuit besar pada QPU yang berisik. Tanpa argumen ini, configuration recovery akan gagal dan menampilkan error jika tidak ada konfigurasi valid yang diberikan.
from functools import partial
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
solve_sci_batch,
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
Iteration 1
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Iteration 2
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Final energy: -108.59275573641656
Final energy error: 0.0032316145694579745
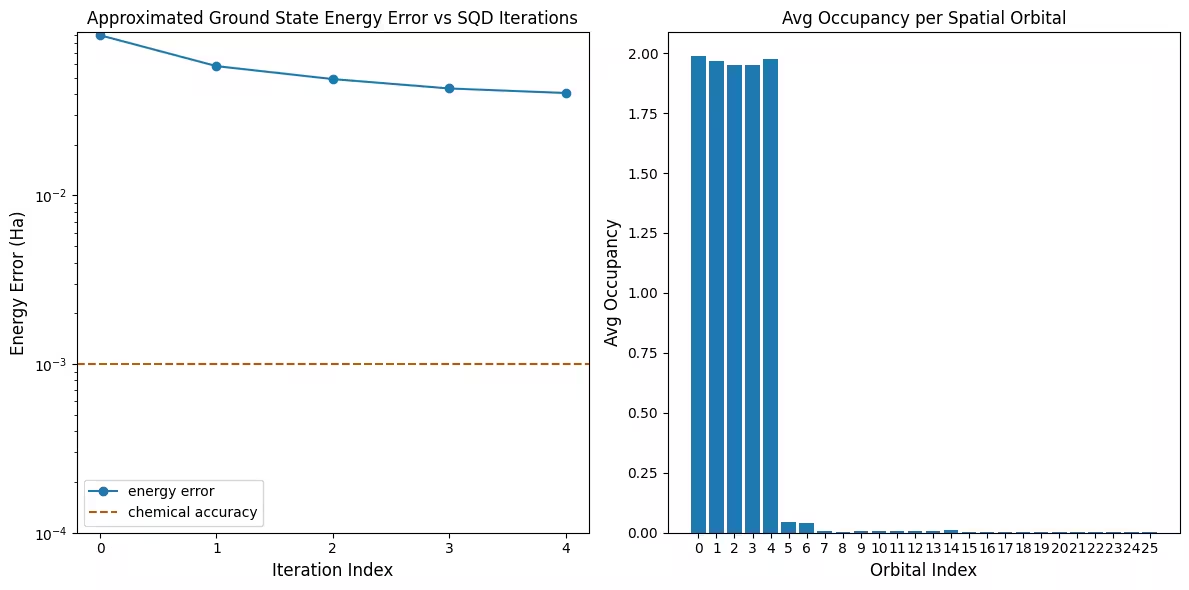
Visualisasikan hasilnya
Plot pertama menunjukkan bahwa dalam simulasi ini kita sudah berada dalam rentang 1 mH dari jawaban eksak setelah iterasi pertama (akurasi kimia umumnya diterima sebagai 1 kcal/mol 1.6 mH). Namun ini adalah sistem kecil, dan karena sampelnya tanpa noise, configuration recovery tidak diperlukan. Pada sistem yang lebih besar yang dijalankan pada QPU yang berisik, beberapa iterasi configuration recovery mungkin diperlukan, dan akurasi akhir mungkin lebih buruk. Secara umum, energi dapat ditingkatkan dengan mengizinkan lebih banyak iterasi configuration recovery atau dengan menambah jumlah sampel per batch.
Plot kedua menunjukkan rata-rata occupancy setiap orbital spasial setelah iterasi terakhir. Kita bisa melihat bahwa elektron spin-up maupun spin-down menempati lima orbital pertama dengan probabilitas tinggi dalam solusi kita.
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()

Contoh hardware skala besar
Sekarang, kita jalankan contoh yang lebih besar pada hardware kuantum nyata. Di sini, kita akan menurunkan ruang aktif untuk molekul nitrogen dari basis set cc-pVDZ.
Langkah 1-4
Di sini kita merangkum semua langkah menjadi satu alur kerja tunggal dalam skala yang lebih besar, yang kemudian dijalankan pada hardware kuantum nyata.
# ------------------------------ Step 1 ------------------------------
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="cc-pvdz",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Store reference energy from SCI calculation performed separately
reference_energy = -109.22802921665716
print(f"norb = {norb}")
print(f"nelec = {nelec}")
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=133
)
print(f"Using backend {backend.name}")
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
# ------------------------------ Step 2 ------------------------------
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
# ------------------------------ Step 3 ------------------------------
sampler = Sampler(mode=backend)
sampler.options.environment.job_tags = ["TUT_SQD"]
job = sampler.run([isa_circuit], shots=100_000)
primitive_result = job.result()
pub_result = primitive_result[0]
# ------------------------------ Step 4 ------------------------------
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the
# orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the
# sci_solver argument in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()
converged SCF energy = -108.929838385609
norb = 26
nelec = (5, 5)
E(CCSD) = -109.2177884185544 E_corr = -0.2879500329450045
Using backend ibm_boston
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20), (24, 24)].
Removing interaction (24, 24) from the end.
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20)].
Removing interaction (20, 20) from the end.
Gate counts: OrderedDict({'sx': 7039, 'rz': 6990, 'cz': 1858, 'x': 61, 'measure': 52, 'barrier': 1})
Fraction of sampled configurations that are valid: 0.02124
Expected fraction of valid configurations from uniformly random bitstrings: 9.607888706852918e-07
Iteration 1
Subsample 0
Energy: -109.13889134249762
Subspace dimension: 120409
Subsample 1
Energy: -109.11785470455858
Subspace dimension: 110889
Subsample 2
Energy: -109.13234360554011
Subspace dimension: 130321
Iteration 2
Subsample 0
Energy: -109.16392179579177
Subspace dimension: 223729
Subsample 1
Energy: -109.16281938332986
Subspace dimension: 223729
Subsample 2
Energy: -109.16955816711932
Subspace dimension: 233289
Iteration 3
Subsample 0
Energy: -109.17905772999075
Subspace dimension: 324900
Subsample 1
Energy: -109.17532445048462
Subspace dimension: 357604
Subsample 2
Energy: -109.1733168689756
Subspace dimension: 348100
Iteration 4
Subsample 0
Energy: -109.18437778820451
Subspace dimension: 474721
Subsample 1
Energy: -109.18450164209159
Subspace dimension: 476100
Subsample 2
Energy: -109.18493571190754
Subspace dimension: 487204
Iteration 5
Subsample 0
Energy: -109.18616522497996
Subspace dimension: 622521
Subsample 1
Energy: -109.18652868888333
Subspace dimension: 644809
Subsample 2
Energy: -109.18753326484406
Subspace dimension: 585225
Final energy: -109.18753326484406
Final energy error: 0.040495951813099396
Langkah selanjutnya
Jika kamu merasa topik ini menarik, kamu mungkin tertarik dengan materi berikut:
- Diagonalisasi kuantum Krylov berbasis sampel untuk model kisi fermionic - tutorial terkait yang menggunakan Circuit evolusi waktu alih-alih ansatz variasional
- Skalakan alur kerja kimia SQD dengan pemecah Dice - halaman yang menunjukkan cara menggunakan perangkat lunak Dice yang lebih efisien untuk diagonalisasi
- Dokumentasi API addon SQD - referensi untuk fungsi
diagonalize_fermionic_hamiltonian - Chemistry beyond the scale of exact diagonalization on a quantum-centric supercomputer - makalah yang menjadi dasar tutorial ini