Meningkatkan estimasi energi Hamiltonian kimia dengan SQD
Dalam tutorial ini kita mengimplementasikan sebuah pola Qiskit yang menunjukkan cara memproses ulang sampel kuantum yang berisik untuk menemukan pendekatan ground state dari Hamiltonian kimia: molekul pada kesetimbangan dalam basis set 6-31G. Kita akan mengikuti pendekatan diagonalisasi kuantum berbasis sampel untuk memproses sampel yang diambil dari ansatz Circuit kuantum 36-Qubit (dalam hal ini, Circuit LUCJ). Untuk memperhitungkan efek noise kuantum, teknik pemulihan konfigurasi digunakan.
Pola ini dapat dijelaskan dalam empat langkah:
- Langkah 1: Pemetaan ke masalah kuantum
- Buat ansatz untuk mengestimasi ground state
- Langkah 2: Optimasi masalah
- Transpile ansatz untuk backend
- Langkah 3: Jalankan eksperimen
- Ambil sampel dari ansatz menggunakan primitif
Sampler
- Ambil sampel dari ansatz menggunakan primitif
- Langkah 4: Proses ulang hasil
- Loop pemulihan konfigurasi self-consistent
- Proses ulang seluruh set sampel bitstring, menggunakan pengetahuan awal tentang jumlah partikel dan rata-rata hunian orbital yang dihitung pada iterasi terbaru.
- Buat kumpulan subsampel secara probabilistik dari bitstring yang telah dipulihkan.
- Proyeksikan dan diagonalisasikan Hamiltonian molekuler pada setiap subruang yang disampel.
- Simpan energi ground state minimum yang ditemukan di semua kumpulan dan perbarui rata-rata hunian orbital.
- Loop pemulihan konfigurasi self-consistent
Untuk contoh ini, Hamiltonian elektron-berinteraksi memiliki bentuk umum:
/ adalah operator kreasi/anihilasi fermionik yang terkait dengan elemen basis set ke- dan spin . dan adalah integral elektronik satu- dan dua-badan.
Alur kerja SQD dengan pemulihan konfigurasi self-consistent digambarkan dalam diagram berikut.
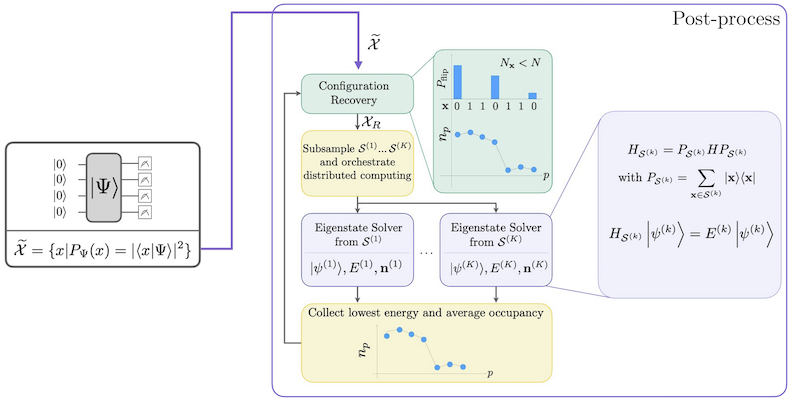
SQD diketahui bekerja dengan baik ketika eigenstate target bersifat jarang: fungsi gelombang didukung dalam satu set state basis yang ukurannya tidak bertambah secara eksponensial seiring ukuran masalah. Dalam skenario ini, diagonalisasi Hamiltonian yang diproyeksikan ke subruang yang didefinisikan oleh :
menghasilkan pendekatan yang baik untuk eigenstate target. Peran perangkat kuantum adalah menghasilkan sampel dari anggota saja. Pertama, sebuah sirkuit kuantum mempersiapkan state di perangkat kuantum. Pengkodean Jordan-Wigner digunakan. Akibatnya, anggota basis komputasi merepresentasikan state Fock (konfigurasi/determinan elektronik). Circuit disampel dalam basis komputasi, menghasilkan set konfigurasi berisik . Konfigurasi direpresentasikan oleh bitstring. Set kemudian dimasukkan ke blok pemrosesan ulang klasik, di mana teknik pemulihan konfigurasi self-consistent digunakan. Dalam kerangka SQD, peran perangkat kuantum adalah menghasilkan distribusi probabilitas.
Langkah 1: Petakan masalah ke Circuit kuantum
Dalam tutorial ini, kita akan mendekati energi ground state molekul . Pertama, kita akan menentukan molekul dan propertinya. Selanjutnya, kita akan membuat ansatz local unitary cluster Jastrow (LUCJ) (Circuit kuantum) untuk menghasilkan sampel dari komputer kuantum guna estimasi energi ground state.
Pertama, kita akan menentukan molekul dan propertinya.
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import warnings
warnings.filterwarnings("ignore")
import pyscf
import pyscf.cc
import pyscf.mcscf
# Specify molecule properties
open_shell = False
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="6-31g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
num_orbitals = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
num_elec_a = (n_electrons + mol.spin) // 2
num_elec_b = (n_electrons - mol.spin) // 2
cas = pyscf.mcscf.CASCI(scf, num_orbitals, (num_elec_a, num_elec_b))
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), num_orbitals)
# Compute exact energy
exact_energy = cas.run().e_tot
converged SCF energy = -108.835236570775
CASCI E = -109.046671778080 E(CI) = -32.8155692383188 S^2 = 0.0000000
Selanjutnya, kita akan membuat ansatz. Ansatz LUCJ adalah Circuit kuantum terparameter, dan kita akan menginisialisasinya dengan amplitudo t2 dan t1 yang diperoleh dari perhitungan CCSD.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -109.0398256929734 E_corr = -0.2045891221988311
Kita akan menggunakan paket ffsim untuk membuat dan menginisialisasi ansatz dengan amplitudo t2 dan t1 yang dihitung di atas. Karena molekul kita memiliki state Hartree-Fock closed-shell, kita akan menggunakan varian spin-balanced dari ansatz UCJ, UCJOpSpinBalanced.
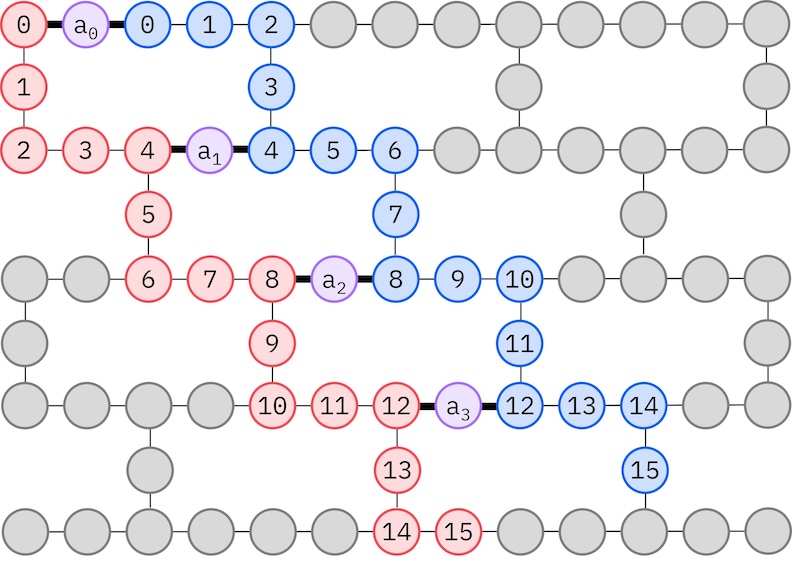
Karena hardware IBM target kita memiliki topologi heavy-hex, kita akan mengadopsi pola zig-zag untuk interaksi qubit. Dalam pola ini, orbital (yang direpresentasikan oleh qubit) dengan spin yang sama terhubung dengan topologi garis (lingkaran merah dan biru) di mana setiap garis mengambil bentuk zig-zag karena konektivitas heavy-hex dari hardware target. Juga, karena topologi heavy-hex, orbital untuk spin yang berbeda memiliki koneksi antara setiap orbital ke-4 (0, 4, 8, dst.) (lingkaran ungu).
import ffsim
from qiskit import QuantumCircuit, QuantumRegister
n_reps = 1
alpha_alpha_indices = [(p, p + 1) for p in range(num_orbitals - 1)]
alpha_beta_indices = [(p, p) for p in range(0, num_orbitals, 4)]
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(alpha_alpha_indices, alpha_beta_indices),
)
nelec = (num_elec_a, num_elec_b)
# create an empty quantum circuit
qubits = QuantumRegister(2 * num_orbitals, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(num_orbitals, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
Langkah 2: Optimasi masalah
Selanjutnya, kita akan mengoptimalkan Circuit kita untuk hardware target. Kita perlu memilih perangkat hardware yang akan digunakan sebelum mengoptimalkan Circuit kita. Kita akan menggunakan fake backend 127-Qubit dari qiskit_ibm_runtime untuk mengemulasi perangkat nyata. Untuk menjalankan pada QPU nyata, cukup ganti fake backend dengan backend nyata. Lihat dokumentasi Qiskit IBM Runtime untuk info lebih lanjut.
from qiskit_ibm_runtime.fake_provider import FakeSherbrooke
backend = FakeSherbrooke()
Selanjutnya, kami merekomendasikan langkah-langkah berikut untuk mengoptimalkan ansatz dan membuatnya kompatibel dengan hardware.
- Pilih qubit fisik (
initial_layout) dari hardware target yang mengikuti pola zig-zag yang dijelaskan di atas. Menyusun qubit dalam pola ini menghasilkan Circuit yang efisien dan kompatibel dengan hardware dengan lebih sedikit Gate. - Buat staged pass manager menggunakan fungsi generate_preset_pass_manager dari Qiskit dengan pilihan
backenddaninitial_layoutkamu. - Set tahap
pre_initdari staged pass manager kamu keffsim.qiskit.PRE_INIT.ffsim.qiskit.PRE_INITmencakup pass Transpiler Qiskit yang mendekomposisi Gate menjadi rotasi orbital dan kemudian menggabungkan rotasi orbital, sehingga menghasilkan lebih sedikit Gate di Circuit akhir. - Jalankan pass manager pada Circuit kamu.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
spin_a_layout = [0, 14, 18, 19, 20, 33, 39, 40, 41, 53, 60, 61, 62, 72, 81, 82]
spin_b_layout = [2, 3, 4, 15, 22, 23, 24, 34, 43, 44, 45, 54, 64, 65, 66, 73]
initial_layout = spin_a_layout + spin_b_layout
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend, initial_layout=initial_layout
)
# without PRE_INIT passes
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/o pre-init passes): {isa_circuit.count_ops()}")
# with PRE_INIT passes
# We will use the circuit generated by this pass manager for hardware execution
pass_manager.pre_init = ffsim.qiskit.PRE_INIT
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/ pre-init passes): {isa_circuit.count_ops()}")
Gate counts (w/o pre-init passes): OrderedDict({'rz': 4420, 'sx': 3432, 'ecr': 1366, 'x': 239, 'measure': 32, 'barrier': 1})
Gate counts (w/ pre-init passes): OrderedDict({'rz': 2460, 'sx': 2156, 'ecr': 730, 'x': 71, 'measure': 32, 'barrier': 1})
Langkah 3: Jalankan eksperimen
Setelah mengoptimalkan Circuit untuk eksekusi hardware, kita siap menjalankannya di hardware target dan mengumpulkan sampel untuk estimasi energi ground state. Karena kita hanya memiliki satu sirkuit, kita akan menggunakan mode eksekusi Job Qiskit Runtime dan mengeksekusi Circuit kita.
Catatan: Kami telah mengomentari kode untuk menjalankan Circuit pada QPU dan meninggalkannya sebagai referensi pengguna. Alih-alih menjalankan pada hardware nyata di panduan ini, kita hanya akan menghasilkan sampel acak yang diambil dari distribusi uniform.
import numpy as np
from qiskit_addon_sqd.counts import generate_bit_array_uniform
# from qiskit_ibm_runtime import SamplerV2 as Sampler
# sampler = Sampler(mode=backend)
# job = sampler.run([isa_circuit], shots=10_000)
# primitive_result = job.result()
# pub_result = primitive_result[0]
# bit_array = pub_result.data.meas
rng = np.random.default_rng(24)
bit_array = generate_bit_array_uniform(10_000, num_orbitals * 2, rand_seed=rng)
Langkah 4: Proses ulang hasil
Sekarang, kita jalankan algoritma SQD menggunakan fungsi diagonalize_fermionic_hamiltonian. Lihat dokumentasi API untuk penjelasan argumen fungsi ini.
Solver yang disertakan dalam addon SQD menggunakan implementasi CI terseleksi dari PySCF, khususnya pyscf.fci.selected_ci.kernel_fixed_space. Contoh di bawah ini juga menunjukkan cara meneruskan argumen kata kunci ke fungsi tersebut melalui solver yang disertakan. Di sini kita meneruskan argumen max_cycle.
from functools import partial
from qiskit_addon_sqd.fermion import SCIResult, diagonalize_fermionic_hamiltonian, solve_sci_batch
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 1
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}")
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=num_orbitals,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -105.45358671756313
Subspace dimension: 5476
Iteration 2
Subsample 0
Energy: -107.95172900082163
Subspace dimension: 249001
Iteration 3
Subsample 0
Energy: -108.97460330369815
Subspace dimension: 339889
Iteration 4
Subsample 0
Energy: -109.02739376648793
Subspace dimension: 440896
Iteration 5
Subsample 0
Energy: -109.030972328451
Subspace dimension: 597529
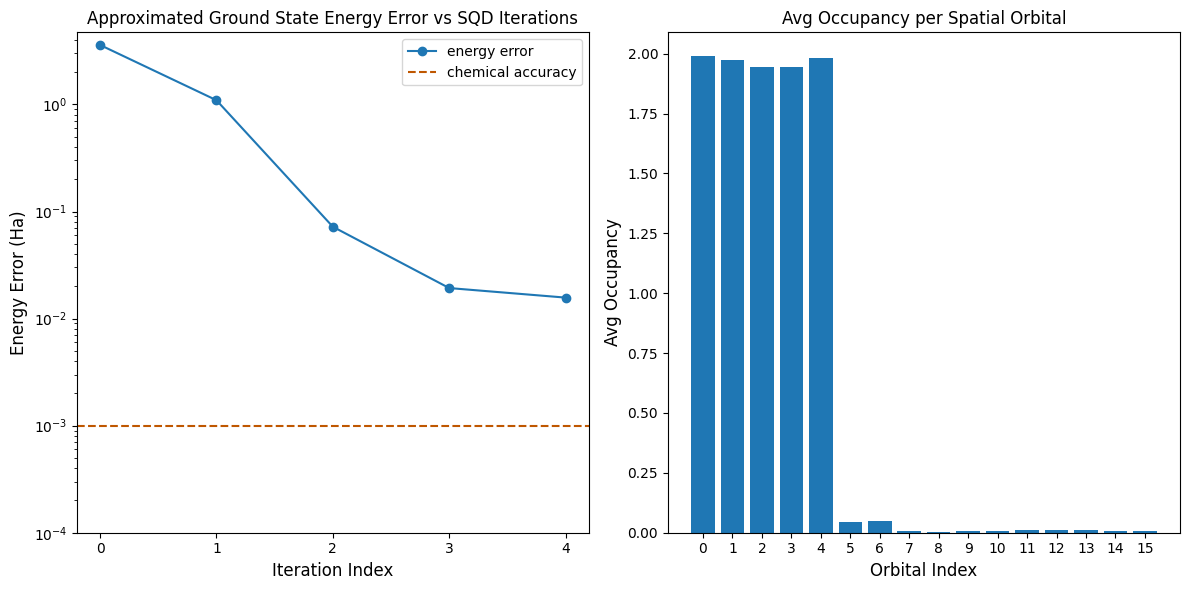
Sekarang, kita plot hasilnya.
Plot pertama menunjukkan bahwa setelah beberapa iterasi kita mengestimasi energi ground state dalam ~16 mH (akurasi kimia biasanya diterima sebagai 1 kcal/mol 1.6 mH). Ingat, sampel kuantum dalam demo ini adalah noise murni. Sinyal di sini berasal dari pengetahuan a priori tentang struktur elektronik dan Hamiltonian molekuler.
Plot kedua menunjukkan rata-rata hunian setiap orbital spasial setelah iterasi terakhir. Kita dapat melihat bahwa elektron spin-up dan spin-down sama-sama menempati lima orbital pertama dengan probabilitas tinggi dalam solusi kita.
import matplotlib.pyplot as plt
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - exact_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(y=chem_accuracy, color="#BF5700", linestyle="--", label="chemical accuracy")
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Exact energy: {exact_energy:.5f} Ha")
print(f"SQD energy: {min_e[-1]:.5f} Ha")
print(f"Absolute error: {e_diff[-1]:.5f} Ha")
plt.tight_layout()
plt.show()
Exact energy: -109.04667 Ha
SQD energy: -109.03097 Ha
Absolute error: 0.01570 Ha