HI-VQE Chemistry - Fungsi Qiskit oleh Qunova Computing
# Added by doQumentation — required packages for this notebook
!pip install -q qiskit-ibm-catalog qiskit-ibm-runtime
# This cell is hidden from users
from qiskit_ibm_runtime import QiskitRuntimeService
service = QiskitRuntimeService()
instance = service.active_account()["instance"]
backend_name = service.least_busy(operational=True, min_num_qubits=16).name
Lihat referensi API
Qiskit Functions adalah fitur eksperimental yang hanya tersedia untuk pengguna IBM Quantum® Premium Plan, Flex Plan, dan On-Prem (via IBM Quantum Platform API) Plan. Fitur ini masih dalam status rilis pratinjau dan dapat berubah sewaktu-waktu.
Versi paket
Kode pada halaman ini dikembangkan menggunakan persyaratan berikut. Kami menyarankan menggunakan versi ini atau yang lebih baru.
qiskit-ibm-runtime~=0.45.0
Gambaran Umum
Dalam kimia kuantum, masalah struktur elektronik berfokus pada pencarian solusi persamaan Schrödinger elektronik — fungsi gelombang kuantum yang menggambarkan perilaku elektron dalam suatu sistem. Fungsi-fungsi gelombang ini adalah vektor amplitudo kompleks, di mana setiap amplitudo merepresentasikan kontribusi dari kemungkinan konfigurasi elektron.
Ground state adalah fungsi gelombang dengan energi terendah dari suatu sistem dan memiliki peran yang sangat penting dalam studi sistem molekuler. Pendekatan paling akurat untuk menghitung ground state mempertimbangkan semua kemungkinan konfigurasi elektron, tetapi ini menjadi tidak praktis untuk sistem yang lebih besar karena jumlah konfigurasi bertumbuh secara eksponensial seiring ukuran sistem.
Handover Iterative Variational Quantum Eigensolver (HI-VQE) adalah metode hibrida kuantum-klasik yang inovatif untuk memperkirakan ground state sistem molekuler secara akurat. Metode ini mengintegrasikan perangkat keras kuantum dengan komputasi klasik, menggunakan prosesor kuantum untuk menjelajahi kandidat konfigurasi elektron secara efisien dan menghitung fungsi gelombang yang dihasilkan pada komputer klasik. Dengan menghasilkan fungsi gelombang yang kompak namun akurat secara kimia, HI-VQE meningkatkan penelitian dan penemuan dalam kimia kuantum dan ilmu material.

HI-VQE mengurangi kompleksitas komputasi masalah struktur elektronik dengan memperkirakan ground state secara efisien dengan akurasi tinggi. Metode ini berfokus pada subset konfigurasi elektron yang paling relevan yang dipilih dengan cermat, mengoptimalkan akurasi dan efisiensi sekaligus.
Dengan menggabungkan kekuatan komputer klasik dan kuantum, HI-VQE secara iteratif memperhalus dan meningkatkan estimasi fungsi gelombang saat ini. Teknik konstruksi subspace uniknya membantu membuat pemilihan konfigurasi lebih efisien, sehingga pengguna memiliki kontrol komputasi yang lebih besar dan akurasi yang lebih baik dalam simulasi kimia kuantum.
Kalau kamu ingin mempelajari algoritma ini lebih dalam, kamu bisa membaca makalah penelitian terkait.
Deskripsi
Jumlah konfigurasi elektron untuk sistem molekuler bertumbuh secara eksponensial seiring ukuran sistem. Namun, untuk keadaan elektronik tertentu, seperti ground state, umum terjadi bahwa hanya sebagian kecil konfigurasi yang berkontribusi signifikan terhadap energi keadaan tersebut. Metode selected configuration interaction (SCI) memanfaatkan kelangkaan ini untuk mengurangi biaya komputasi dengan mengidentifikasi dan berfokus pada konfigurasi yang paling relevan. Subset konfigurasi ini disebut sebagai subspace.
HI-VQE memanfaatkan efisiensi inheren komputer kuantum dalam merepresentasikan sistem molekuler untuk membantu pencarian subspace. Metode ini mengintegrasikan subrutin klasik dan kuantum untuk memecahkan masalah struktur elektronik dengan akurasi tinggi. Berbeda dengan metode SCI kuantum yang sudah ada, HI-VQE menggabungkan pelatihan variasional, konstruksi subspace iteratif, dan penyaringan konfigurasi pre-diagonalization untuk meningkatkan efisiensi dengan mengurangi pengukuran kuantum, iterasi, dan biaya diagonalisasi klasik. HI-VQE oleh karena itu dapat diterapkan pada sistem molekuler yang lebih besar yang membutuhkan lebih banyak qubit, dan mengurangi biaya untuk memecahkan masalah dengan ukuran tertentu hingga tingkat akurasi yang sama.
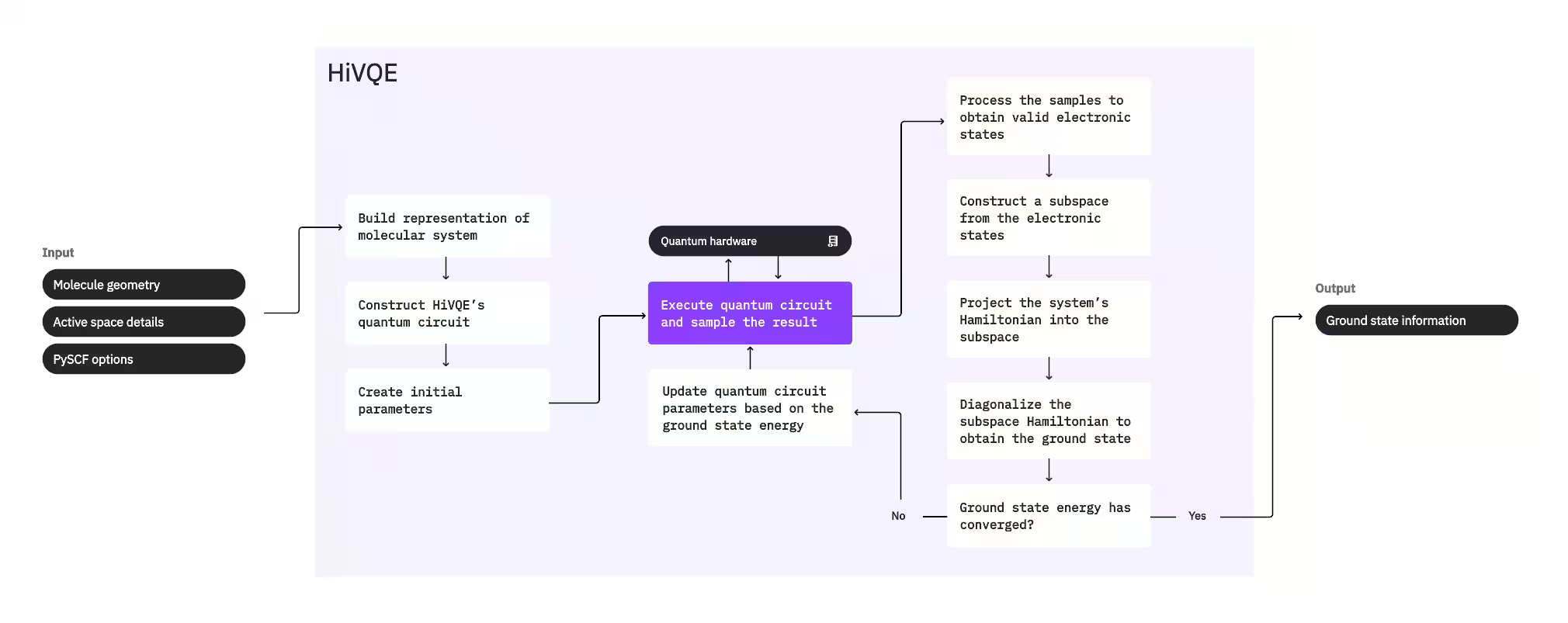
Untuk menghitung ground state suatu sistem, HI-VQE pertama-tama menggunakan paket kimia klasik PySCF untuk menghasilkan representasi molekuler dari input yang diberikan pengguna, seperti geometri molekuler dan informasi molekuler lainnya. Kemudian masuk ke dalam loop optimasi hibrida kuantum-klasik, secara iteratif memperhalus subspace untuk merepresentasikan ground state secara optimal sambil meminimalkan jumlah konfigurasi yang disertakan. Loop berlanjut sampai kriteria konvergensi, seperti ukuran subspace atau stabilitas energi, terpenuhi, setelah itu fungsi gelombang dan energi ground state yang telah dihitung dikeluarkan. Hasil ini dapat digunakan untuk membangun permukaan energi potensial yang akurat dan melakukan analisis lebih lanjut dari sistem tersebut.
Loop optimasi berfokus pada penyesuaian parameter Circuit kuantum untuk menghasilkan subspace berkualitas tinggi. HI-VQE menawarkan tiga opsi Circuit kuantum: excitation_preserving, efficient_su2, dan LUCJ. Optimasi diinisialisasi dekat dengan keadaan referensi Hartree-Fock karena kesesuaiannya yang umum. Circuit kemudian dieksekusi pada perangkat kuantum dan konfigurasi disampel dari keadaan kuantum yang dihasilkan sebelum dikembalikan sebagai string biner. Karena noise perangkat kuantum, beberapa konfigurasi yang disampel mungkin tidak valid secara fisik, gagal menjaga jumlah elektron atau spin. HI-VQE mengatasi ini menggunakan proses pemulihan konfigurasi dari paket qiskit-addon-sqd, sehingga pengguna dapat mengoreksi konfigurasi yang tidak valid atau membuangnya.
Konfigurasi yang valid kemudian menjalani langkah penyaringan opsional untuk menghapus konfigurasi yang diprediksi berkontribusi minimal. Ini mengurangi dimensi subspace, sehingga menurunkan biaya langkah diagonalisasi. Jika penyaringan diaktifkan, maka Hamiltonian subspace awal dikonstruksi dari konfigurasi yang valid dan diagonalisasi dilakukan dengan kriteria penghentian yang sangat longgar. Meskipun akurasi amplitudo yang dihasilkan untuk setiap konfigurasi rendah, cara ini efektif untuk memprediksi konfigurasi mana yang harus dikeluarkan dari subspace pada iterasi ini, dan cepat untuk dihitung.
Konfigurasi yang dipilih ditambahkan ke subspace, dan Hamiltonian sistem diproyeksikan ke dalam subspace ini. Subspace diperbarui secara iteratif, mempertahankan konfigurasi yang paling relevan di seluruh iterasi. Pendekatan ini kontras dengan metode alternatif karena Circuit kuantum tidak perlu memperkirakan keseluruhan ground state di setiap langkah.
Selanjutnya, Hamiltonian subspace didiagonalisasi secara klasik untuk mendapatkan eigenvalue terendah dan eigenvector yang sesuai, merepresentasikan perkiraan ground state dan energinya. Saat kualitas subspace meningkat melalui iterasi, ground state yang dihitung semakin mendekati ground state sebenarnya. Langkah penyaringan tambahan dapat dilakukan pada titik ini untuk menghapus konfigurasi dari subspace yang tidak memiliki kontribusi substansial terhadap ground state yang dihitung. Langkah ini memastikan bahwa subspace yang dibawa ke iterasi berikutnya sekompak mungkin. Ini dievaluasi berdasarkan amplitudo yang dikembalikan oleh diagonalisasi, karena ini merepresentasikan kontribusi kepentingan setiap konfigurasi terhadap ground state yang dihitung.
Pemeriksaan konvergensi kemudian menentukan apakah pelatihan lebih lanjut akan meningkatkan hasil. Jika ya, maka langkah ekspansi klasik opsional dilakukan, parameter Circuit kuantum diperbarui untuk lebih meminimalkan energi yang dihitung, dan proses berulang. Langkah ekspansi klasik menghasilkan konfigurasi tambahan untuk subspace, melengkapi konfigurasi yang disampel dari perangkat kuantum. Pertama-tama mengidentifikasi konfigurasi dengan amplitudo terbesar dalam hasil diagonalisasi, sebelum menghasilkan konfigurasi baru dengan eksitasi tunggal dan ganda dari konfigurasi yang diidentifikasi. Jumlah konfigurasi yang diinginkan kemudian ditambahkan ke subspace.
Setelah ditentukan bahwa iterasi telah konvergen, HI-VQE mengembalikan ground state yang dihitung (dalam bentuk keadaan dalam subspace dan amplitudunya dalam fungsi gelombang ground state), energinya, dan ukuran variansi energi yang memberikan indikasi apakah keadaan yang dihitung membentuk eigenstate dari Hamiltonian sistem.
Pengguna dapat memutuskan Circuit kuantum yang digunakan dan jumlah shot yang diambil untuk setiap sirkuit kuantum, serta mengontrol ukuran subspace atau mengaktifkan generasi klasik konfigurasi tambahan untuk membantu konfigurasi yang dihasilkan kuantum. Dengan cara ini pengguna dapat menyesuaikan perilaku HI-VQE sesuai dengan aplikasi yang mereka inginkan.
Lisensi
Perlu diperhatikan bahwa penggunaan Qiskit Function ini terbatas pada masalah yang memerlukan paling banyak 20 qubit, kecuali jika lisensi diperoleh yang memberikan batas lebih tinggi.
Silakan kirim email ke qiskit.support@qunovacomputing.com jika kamu ingin menanyakan tentang cara memperoleh lisensi.
Memulai
Pertama, minta akses ke fungsi ini. Kemudian, autentikasi menggunakan API key IBM Quantum® kamu dan, dengan asumsi kamu sudah menyimpan akun ke lingkungan lokalmu, pilih Qiskit Function sebagai berikut:
import reprlib
from qiskit_ibm_catalog import QiskitFunctionsCatalog
catalog = QiskitFunctionsCatalog(channel="ibm_quantum_platform")
function = catalog.load("qunova/hivqe-chemistry")
Contoh
Contoh pertama menunjukkan cara menghitung energi ground state molekul NH3 menggunakan algoritma HI-VQE.
Mendefinisikan geometri molekuler dan opsi
Geometri molekuler NH3 diberikan dengan koordinat Kartesian yang dipisahkan dengan ";" untuk setiap atom.
# Define the molecule geometry
geometry = """
N -0.85188 -0.02741 0.03141;
H 0.16545 0.00593 -0.01648;
H -1.16348 -0.39357 -0.86702;
H -1.16348 0.94228 0.06281;
"""
Opsi tambahan dapat didefinisikan dan disediakan untuk sistem molekuler dalam format kamus berikut.
# Configure some options for the job.
molecule_options = {"basis": "sto3g"}
hivqe_options = {"shots": 100, "max_iter": 20}
Eksekusi fungsi dengan input geometri dan opsi.
# Run HI-VQE
job = function.run(
geometry=geometry,
# `backend_name` is the name of a backend with at least 16 qubits,
# for example, "ibm_marrakesh".
backend_name=backend_name,
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
Sebaiknya cetak ID job Fungsi agar dapat diberikan dalam permintaan dukungan jika ada yang tidak beres.
print("Job ID:", job.job_id)
Job ID: e5ced6f2-fd1d-4244-a6aa-bd27cfb0cdee
Contoh ini kemudian menggunakan 16 qubit dengan 8 orbital basis sto3g untuk molekul NH3. Periksa status beban kerja Qiskit Function kamu atau ambil hasil sebagai berikut:
print(job.status())
QUEUED
Setelah job selesai, hasilnya dapat diperoleh dengan instance result().
result = job.result()
# Output can be long, so we display a shortened representation
shortened_result = reprlib.repr(result)
print(shortened_result)
{'eigenvector': [0.9824448589364075, 0.009527106392132133, 6.854074372058527e-08, 3.591500190038039e-07, 0.0012975231577544268, 2.310159709002111e-05, ...], 'energy': -55.52108557170985, 'energy_history': [-55.51901898989887, -55.52056881448526, -55.52065046778772, -55.520690696813716, -55.520691108428, -55.520708448092634, ...], 'energy_variance': 3.066239097617371e-10, ...}
Untuk mengakses energi ground state, gunakan kunci "energy". Kunci "eigenvector" memberikan koefisien CI dengan notasi bitstring yang sesuai dari konfigurasi elektron yang tersimpan dengan "states" dari hasilnya.
fci_energy = -55.521148034704126 # the exact energy using FCI method
hivqe_energy = result["energy"]
print(
f"|Exact Energy - HI-VQE Energy|: "
f"{abs(fci_energy - hivqe_energy) * 1000} mHa"
)
print(f"Sampled Number of States: {len(result['states'])}")
|Exact Energy - HI-VQE Energy|: 0.06246299427914437 mHa
Sampled Number of States: 1936
Performa
Bagian ini menunjukkan perhitungan benchmark yang telah didemonstrasikan dari HI-VQE dengan kasus 24-qubit untuk Li2S, kasus 40-qubit untuk molekul N2, dan kasus 44-qubit untuk sistem FeP-NO.
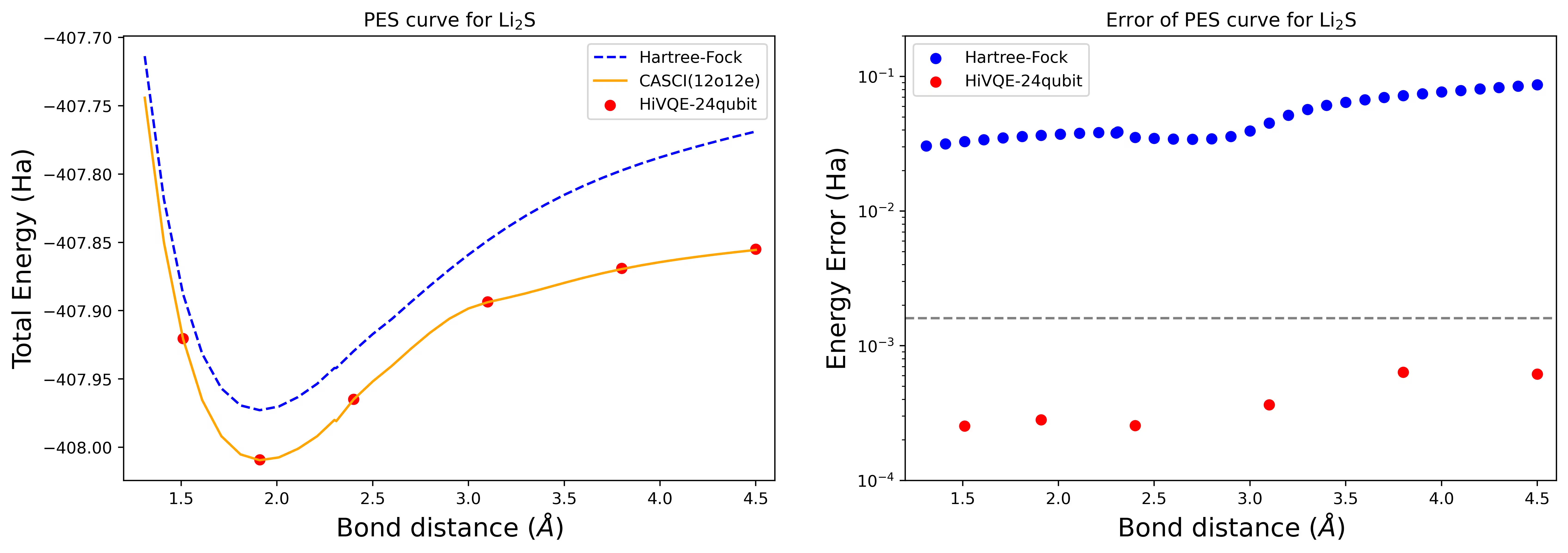
Kurva permukaan energi potensial disosiasi untuk molekul Li2S dengan 24 qubit
Kurva PES ditampilkan dengan referensi FCI dan tebakan awal dari RHF, bersama dengan kesalahan energi dari referensi FCI.
 .
.
Perhitungan telah dilakukan dengan geometri dan opsi berikut.
# This cell is hidden from users
backend_name = service.least_busy(operational=True, min_num_qubits=38).name
# Define Li2S geometries
Li2S_geoms = {
"Li2S_1.51": "S -1.239044 0.671232 -0.030374;Li -1.506327 0.432403 -1.498949;Li -0.899996 0.973348 1.826768;",
"Li2S_2.40": "S -1.741432 0.680397 0.346702;Li -0.529307 0.488006 -1.729343;Li -1.284307 0.989409 2.177209;",
"Li2S_3.80": "S -2.707255 0.674298 0.909161;Li 0.079218 0.552012 -1.671656;Li -0.927010 0.931502 1.557063;",
}
# Configure some options for the job.
molecule_options = {
"basis": "sto3g",
}
hivqe_options = {
"shots": 100,
"max_iter": 20,
}
results = []
for geom in ["Li2S_1.51", "Li2S_2.40", "Li2S_3.80"]:
# Run HI-VQE
job = function.run(
geometry=Li2S_geoms[geom],
backend_name=backend_name, # can use any device with at least 38 qubits
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
results.append(job.result())
Titik merah merepresentasikan hasil perhitungan HI-VQE untuk enam geometri berbeda, dan tiga geometri yang bersesuaian dengan 1,51, 2,40, dan 3,80 Angstrom disediakan sebagai input dalam sel di atas.
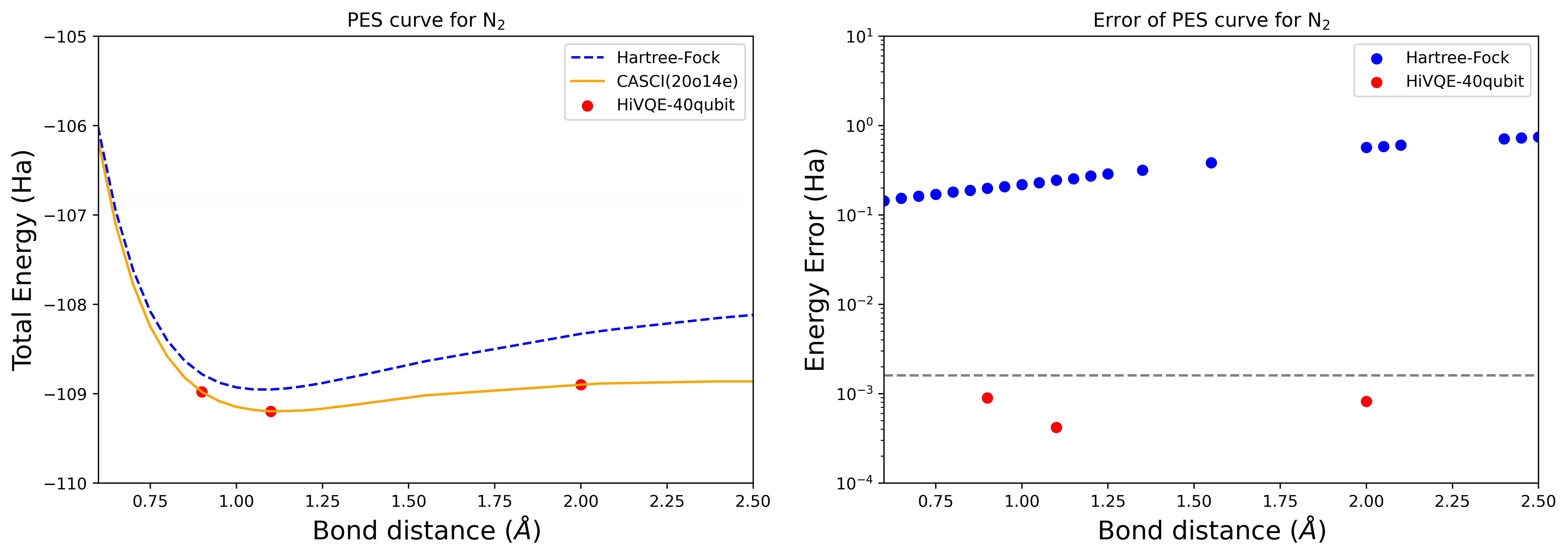
Kurva PES disosiasi untuk molekul N2 dengan 40 qubit
Molekul nitrogen telah diidentifikasi sebagai sistem multireferensi dengan kontribusi energi korelasi yang besar melampaui keadaan Hartree-Fock. Kami melakukan perhitungan benchmark untuk molekul N2 dengan basis cc-pvdz, (20o,14e) menggunakan pemilihan orbital aktif homo-lumo. Jumlah complete active space (CAS) untuk merepresentasikan masalah ini adalah 6.009.350.400. Tidak mungkin mendapatkan solusi masalah nilai eigen (untuk energi dan struktur elektronik) dengan jumlah keadaan ini menggunakan komputer desktop yang powerful (16cpu/64GB). Dengan HI-VQE, pengguna dapat secara efisien mencari subspace keadaan CAS untuk menemukan hasil yang akurat secara kimia sambil menghemat sumber daya komputasi secara signifikan. Plot berikut menunjukkan kurva PES dari perhitungan HI-VQE 40-qubit untuk disosiasi molekul N2.
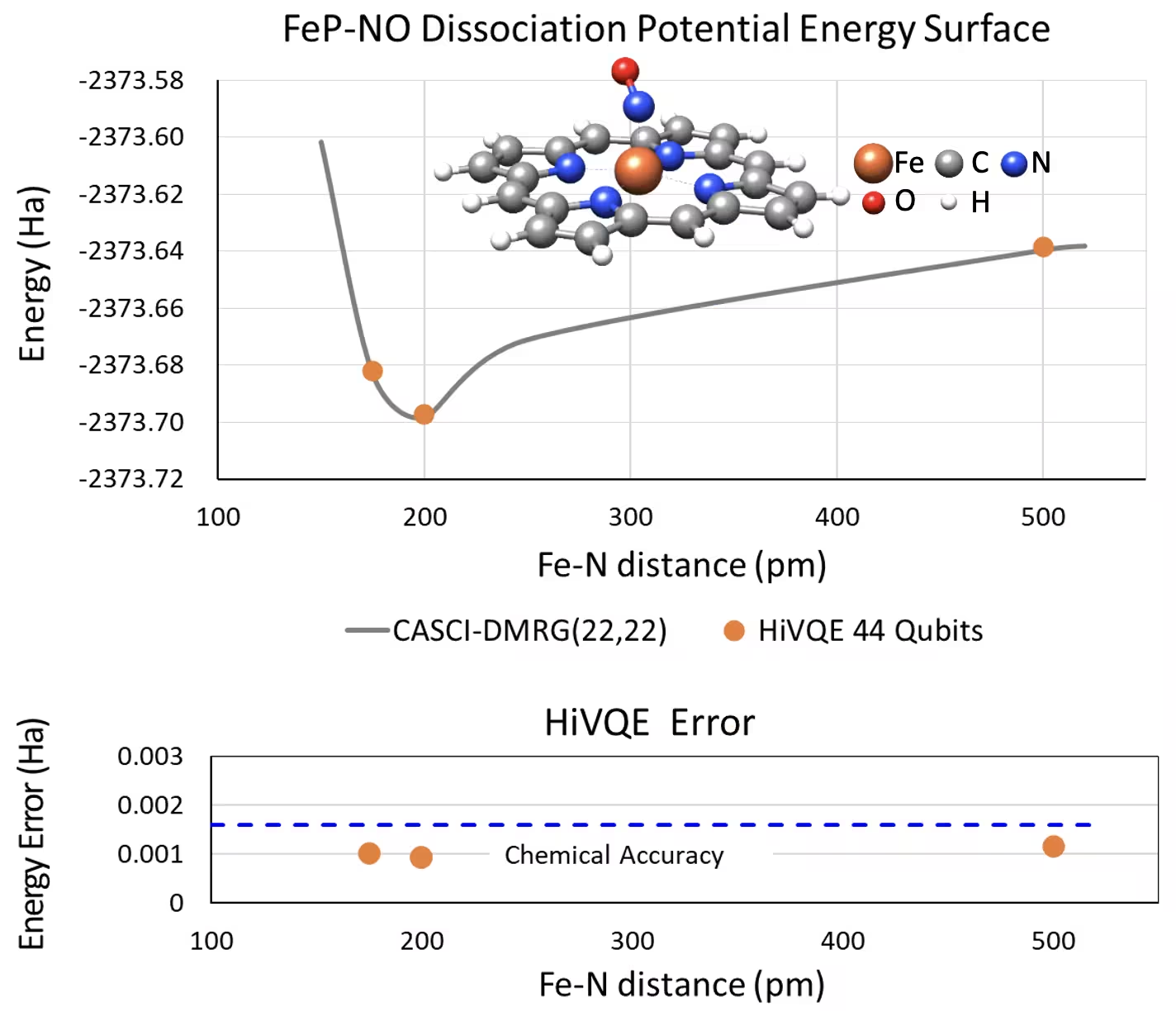
Kurva PES disosiasi untuk iron(II)-porphyrin berkoordinasi lima dengan sistem NO dengan 44 qubit
Sistem kimia menarik lainnya adalah kompleks iron(II)-porphyrin (FeP) dengan ligan nitric oxide (NO) yang terkoordinasi, yang merepresentasikan sistem metaloporfirin yang relevan secara biologis dan memainkan peran penting dalam berbagai proses fisiologis. Dalam contoh ini, HI-VQE telah digunakan untuk memperkirakan kurva permukaan energi potensial yang akurat dari interaksi antarmolekul antara FeP dan NO (energi ground state untuk geometri yang berbeda-beda jaraknya). Sistem gabungan memiliki 450 orbital dan 202 elektron (450o,202e) dengan basis 6-31g(d) secara total. Pemilihan orbital aktif homo-lumo digunakan untuk menghitung kasus yang lebih kecil dari kasus nyata dengan (22o,22e). Dari hasil benchmark berikut, kami mampu mencapai akurasi kimia (> 1,6 mHa) dengan perhitungan kimia komputer klasik mutakhir dari CASCI(DMRG) (22o,22e) sebagai referensi.
Benchmark
- Ukuran matriks eksak adalah jumlah determinan untuk solusi eksak, seperti FCI dan CASCI.
- Perhitungan HI-VQE mengambil sampel dan menghitung subspace dari itu (yaitu, ukuran matriks HI-VQE).
- Total waktu mencakup runtime QPU dan eksekusi Qiskit Function dengan CPU.
- Akurasi diperkirakan dari perbedaan energi dari solusi eksak.
| Sistem kimia | Jumlah qubit | Ukuran matriks eksak | Ukuran matriks HI-VQE | E(diff) dari eksak (mHa) | Jumlah iterasi | Total waktu | Penggunaan runtime QPU |
|---|---|---|---|---|---|---|---|
| (8o,10e) | 16 | 3136 | 1936 | 0.08 | 6 | 37 s | 34 s |
| (10o,10e) | 20 | 63504 | 3969 | 0.60 | 5 | 250 s | 50 s |
| (15o,10e) | 30 | 9018009 | 49729 | 0.90 | 5 | 354 s | 54 s |
| (16o,14e) | 32 | 130873600 | 1798281 | 1.10 | 9 | 6531 s | 121 s |
| (18o,24e) | 36 | 344622096 | 399424 | 0.90 | 24 | 5174 s | 130 s |
| (20o,14e) | 40 | 6009350400 | 9012004 | 1.20 | 21 | 46547 s | 258 s |
Mengambil pesan error
Jika beban kerjamu gagal, statusnya akan menjadi ERROR dan memanggil job.result() akan memunculkan pengecualian:
job = function.run(
geometry="invalid-geometry", # This will cause an error
backend_name=backend_name,
max_states=2000,
max_expansion_states=15,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
job.result()
job.status()
'ERROR'
Mendapatkan dukungan
Kamu bisa mengirim email ke qiskit.support@qunovacomputing.com untuk mendapatkan bantuan dengan fungsi ini.
Jika kamu butuh bantuan untuk mengatasi error tertentu, tolong sertakan ID job Fungsi dari job yang mengalami error tersebut.
Langkah selanjutnya
- Minta akses ke fungsi dengan mengisi formulir ini.
- Kunjungi referensi API untuk Qiskit Function ini.
- Coba tutorial Compute dissociation PES curve for FeP-NO with HI-VQE.
- Tinjau Pellow-Jarman, A., et al. (2025). HIVQE: Handover Iterative Variational Quantum Eigensolver for Efficient Quantum Chemistry Calculations. arXiv preprint arXiv:2503.06292.
- Coba tutorial Dissociation PES curves with Qunova HiVQE.