Variational Quantum Eigensolver (VQE)
Untuk modul ini, siswa harus memiliki lingkungan Python yang berfungsi, dan versi terbaru dari paket-paket berikut yang sudah terinstal:
qiskitqiskit_ibm_runtimeqiskit-aerqiskit.visualizationnumpypylatexenc
Untuk menyiapkan dan menginstal paket-paket ini, lihat panduan Install Qiskit. Untuk menjalankan job di komputer kuantum nyata, siswa perlu menyiapkan akun IBM Cloud, mengikuti langkah-langkah di panduan Set up your IBM Cloud account.
Modul ini telah diuji dan menggunakan sekitar 8 menit waktu QPU. Ini adalah perkiraan, dan penggunaan aktualmu mungkin berbeda.
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-aer qiskit-ibm-runtime scipy
# Uncomment and modify this line as needed to install dependencies
#!pip install 'qiskit>=2.1.0' 'qiskit-ibm-runtime>=0.40.1' 'qiskit-aer>=0.17.0' 'numpy' 'pylatexenc'
Pendahuluan
Sejak pengembangan model mekanika kuantum pada awal abad ke-20, para ilmuwan telah memahami bahwa elektron tidak mengikuti jalur tetap di sekitar inti atom, melainkan berada di wilayah probabilitas yang disebut orbital. Orbital-orbital ini berhubungan dengan tingkat energi diskret tertentu yang dapat ditempati elektron. Elektron secara alami menempati tingkat energi terendah yang tersedia, yang dikenal sebagai keadaan dasar (ground state). Namun, jika sebuah elektron menyerap energi yang cukup, ia bisa melompat ke tingkat energi yang lebih tinggi, memasuki keadaan tereksitasi (excited state). Keadaan tereksitasi ini bersifat sementara, dan elektron pada akhirnya akan kembali ke tingkat energi yang lebih rendah, melepaskan energi yang diserap, sering kali dalam bentuk cahaya. Proses fundamental penyerapan dan emisi energi ini penting untuk memahami bagaimana atom berinteraksi dan membentuk ikatan.
Ketika atom-atom bergabung membentuk molekul, orbital atom mereka bergabung membentuk orbital molekul. Susunan dan tingkat energi elektron dalam orbital molekul ini menentukan sifat molekul yang dihasilkan dan kekuatan ikatan kimia. Misalnya, dalam pembentukan molekul hidrogen () dari dua atom hidrogen individual, elektron dari masing-masing atom menempati orbital atom. Saat atom-atom saling mendekati, orbital atom ini tumpang tindih dan bergabung membentuk orbital molekul baru — satu dengan energi lebih rendah (orbital ikatan/bonding orbital) dan satu dengan energi lebih tinggi (orbital anti-ikatan/anti-bonding orbital). Dua elektron, satu dari masing-masing atom hidrogen, akan lebih memilih menempati orbital ikatan berenergi lebih rendah, yang mengarah pada pembentukan ikatan kovalen stabil yang menyatukan molekul . Perbedaan energi antara atom-atom yang terpisah dan molekul yang terbentuk, khususnya energi elektron dalam orbital molekul, menentukan stabilitas dan sifat ikatan.
Pada bagian berikut, kita akan menjelajahi proses pembentukan molekul ini, dengan fokus pada molekul . Kita akan menggunakan komputer kuantum nyata, dikombinasikan dengan teknik optimasi klasik, untuk menemukan energi dari proses sederhana namun fundamental ini. Eksperimen ini akan memberikan demonstrasi praktis tentang bagaimana komputasi kuantum dapat diterapkan untuk memecahkan masalah dalam kimia komputasional, memberikan wawasan tentang peran energi elektron.
VQE - Algoritma kuantum variasional untuk masalah nilai eigen
Teknik aproksimasi untuk kimia - prinsip variasional dan basis set
Kontribusi Erwin Schrödinger pada mekanika kuantum tidak terbatas pada memperkenalkan model elektronik baru; secara fundamental, ia menegakkan mekanika gelombang dengan mengembangkan persamaan Schrödinger terkenal yang bergantung waktu:
Di sini, adalah operator Hamiltonian, yang mewakili total energi sistem, dan adalah fungsi gelombang yang berisi semua informasi tentang keadaan kuantum sistem. (Catatan: adalah turunan total terhadap waktu, dan kita tidak secara eksplisit menyertakan nilai eigen energi di sini.)
Namun, dalam banyak aplikasi praktis — seperti menentukan tingkat energi yang diizinkan dari atom dan molekul — kita justru menggunakan persamaan Schrödinger tak bergantung waktu (persamaan nilai eigen energi), yang diturunkan dari bentuk bergantung waktu dengan mengasumsikan keadaan stasioner. Keadaan stasioner adalah keadaan kuantum di mana kerapatan probabilitas untuk menemukan partikel di titik tertentu dalam ruang tidak berubah seiring waktu.
Dalam bentuk ini, mewakili nilai eigen energi yang berhubungan dengan keadaan kuantum . Hamiltonian mencakup berbagai kontribusi energi, seperti energi kinetik elektron dan inti, gaya tarik antara elektron dan inti, dan gaya tolak antara elektron.
Menyelesaikan persamaan nilai eigen energi memungkinkan kita menghitung tingkat energi terkuantisasi dari sistem atom dan molekul. Namun, untuk molekul, menyelesaikannya secara eksak sulit karena fungsi gelombang , yang menggambarkan distribusi spasial elektron, bersifat kompleks dan berdimensi tinggi.
Akibatnya, para ilmuwan menggunakan teknik aproksimasi untuk mendapatkan solusi yang praktis dan akurat. Dalam karya ini, kita akan fokus pada dua metode utama:
-
Prinsip variasional
Metode ini mengaproksimasi fungsi gelombang dan menyesuaikannya agar sedekat mungkin dengan energi target, biasanya energi ground state sistem. Ide utama di balik prinsip variasional sangat sederhana:
- Jika kita menebak fungsi gelombang (sebuah "fungsi trial"), energi yang dihitung darinya akan selalu sama dengan atau lebih tinggi dari energi ground state () sistem.
- Dengan menyesuaikan parameter dalam fungsi trial, , kita dapat mendapatkan aproksimasi energi ground state yang semakin baik.
- Akurasinya sangat bergantung pada pilihan fungsi gelombang trial . Fungsi trial yang dipilih dengan buruk dapat menghasilkan estimasi energi yang jauh dari akurat.
-
Aproksimasi basis set
Metode aproksimasi kedua hadir pada tahap konstruksi fungsi gelombang — pendekatan basis set. Dalam kimia kuantum, menyelesaikan persamaan Schrödinger secara eksak untuk molekul hampir tidak mungkin. Sebagai gantinya, kita mengaproksimasi fungsi gelombang multi-elektron yang kompleks dengan membangunnya dari fungsi matematis yang lebih sederhana dan telah ditentukan. Basis set pada dasarnya adalah kumpulan fungsi matematis yang diketahui ini, biasanya dipusatkan pada atom-atom dalam molekul, yang digunakan sebagai blok pembangun untuk merepresentasikan bentuk dan perilaku elektron dalam sistem. Bayangkan seperti mencoba membuat ulang sebuah patung detail menggunakan hanya koleksi balok LEGO standar — semakin banyak jenis dan ukuran balok yang kamu miliki (semakin besar basis setnya), semakin akurat kamu dapat mengaproksimasi bentuk aslinya.
Fungsi basis ini sering terinspirasi dari solusi analitik untuk sistem sederhana seperti atom hidrogen, mengambil bentuk seperti fungsi tipe Gaussian atau Slater, meskipun keduanya masih merupakan aproksimasi. Alih-alih bekerja dengan orbital molekul penuh yang secara teoritis "eksak" namun tidak dapat diatasi, kita mengekspresikannya sebagai kombinasi linier (jumlah dengan koefisien) dari fungsi basis ini. Metode ini dikenal sebagai pendekatan Linear Combination of Atomic Orbitals (LCAO) ketika fungsi basis menyerupai orbital atom. Dengan mengoptimalkan koefisien dalam kombinasi linier ini, kita dapat menemukan fungsi gelombang aproksimasi dan energi terbaik yang mungkin dalam keterbatasan basis set yang dipilih.
- Semakin banyak fungsi yang dimasukkan dalam basis set, semakin baik aproksimasi, tetapi ini datang dengan biaya upaya komputasi yang lebih tinggi.
- Basis set kecil memberikan estimasi kasar, sementara basis set besar memberikan hasil yang lebih presisi dengan mengorbankan sumber daya komputasi yang lebih banyak.
Singkatnya, untuk membuat perhitungan layak dilakukan dan mengurangi biaya komputasi, kita menggunakan prinsip variasional dengan mengaproksimasi fungsi gelombang, yang mengurangi kompleksitas komputasi dan memungkinkan optimasi iteratif untuk meminimalkan energi. Sementara itu, pendekatan basis set menyederhanakan perhitungan dengan merepresentasikan orbital atom sebagai kombinasi fungsi yang telah ditentukan, daripada menyelesaikan fungsi gelombang kontinu secara langsung.
Cek pemahamanmu
Pertimbangkan fungsi gelombang trial di mana adalah konstanta normalisasi dan adalah parameter yang dapat disesuaikan.
(a) Normalisasi fungsi gelombang trial dengan menentukan sedemikian sehingga .
Jawaban
Untuk menormalisasi fungsi gelombang trial yang diberikan:
Gunakan integral Gaussian:
set kemudian dapatkan:
(b) Hitung nilai ekspektasi Hamiltonian yang diberikan oleh di mana , yang berhubungan dengan potensial osilator harmonik sederhana.
Jawaban
Hamiltonian untuk osilator harmonik adalah:
Nilai ekspektasi energi kinetik
Mengambil turunan kedua:
Sehingga:
Menggunakan hasil integral Gaussian standar:
Nilai ekspektasi energi potensial
Menggunakan:
kita dapatkan:
Nilai ekspektasi energi total
(c) Gunakan prinsip variasional untuk menemukan optimal dengan meminimalkan .
Jawaban
Optimasi untuk energi minimum
Diferensiasi
Menyelesaikan:
Substitusi ke :
yang cocok dengan energi ground state osilator harmonik kuantum eksak.
VQE (Variational Quantum Eigensolver)
Variational quantum eigensolver (VQE) adalah metode utama yang akan kita gunakan untuk mengeksplorasi proses , dan di sini, kita akan melihat apa itu VQE dan bagaimana cara kerjanya. Tapi pertama-tama mari berhenti sebentar dan lihat satu hal yang sangat penting melalui pertanyaan check-in berikut.
Cek pemahamanmu
Jika kita sudah memiliki begitu banyak strategi untuk masalah kimia, lalu mengapa kita membutuhkan komputer kuantum? Dan apa tujuan menggunakan komputer kuantum dan klasik bersama-sama?
Jawaban
Komputasi kuantum punya kesempatan untuk merevolusi kimia dengan menangani masalah yang sulit diselesaikan komputer klasik karena skala eksponensial dari keadaan kuantum. Richard Feynman dengan terkenal mencatat bahwa untuk mensimulasikan alam, komputasi juga harus bersifat kuantum [ref 1].
Misalnya, mensimulasikan kafein dengan basis set paling sederhana (STO-3G) akan membutuhkan bit, jauh lebih besar dari jumlah total bintang di alam semesta yang dapat diamati () [ref 2]. Sebuah komputer kuantum dapat mendeskripsikan orbital elektronik kafein dengan 160 qubit.
Komputer kuantum secara alami memproses interaksi kuantum menggunakan superposisi dan entanglement, yang memberikan cara menjanjikan untuk memungkinkan simulasi molekul yang akurat. Lebih jauh, kita dapat menggabungkan keunggulan komputer kuantum (simulasi elektron) dan komputer klasik (pra/pasca-pemrosesan data, manajemen proses algoritma, optimasi, dan sebagainya). Ini diharapkan dapat meningkatkan penemuan material, desain obat, dan prediksi reaksi, mengurangi eksperimen trial-and-error yang mahal. [ref 3][ref 4]
Jika kamu ingin tahu mengapa komputer kuantum dibutuhkan untuk masalah kimia dan mengapa menggunakan sumber daya komputasi kuantum dan klasik bersama-sama, cek artikel-artikel berikut:
Sekarang mari kembali ke VQE.
VQE menggabungkan kekuatan komputer kuantum dengan komputer klasik, secara fundamental menggunakan prinsip variasional untuk mendapatkan energi ground state sistem. Untuk memahami VQE, pertama-tama pecah menjadi tiga bagian:
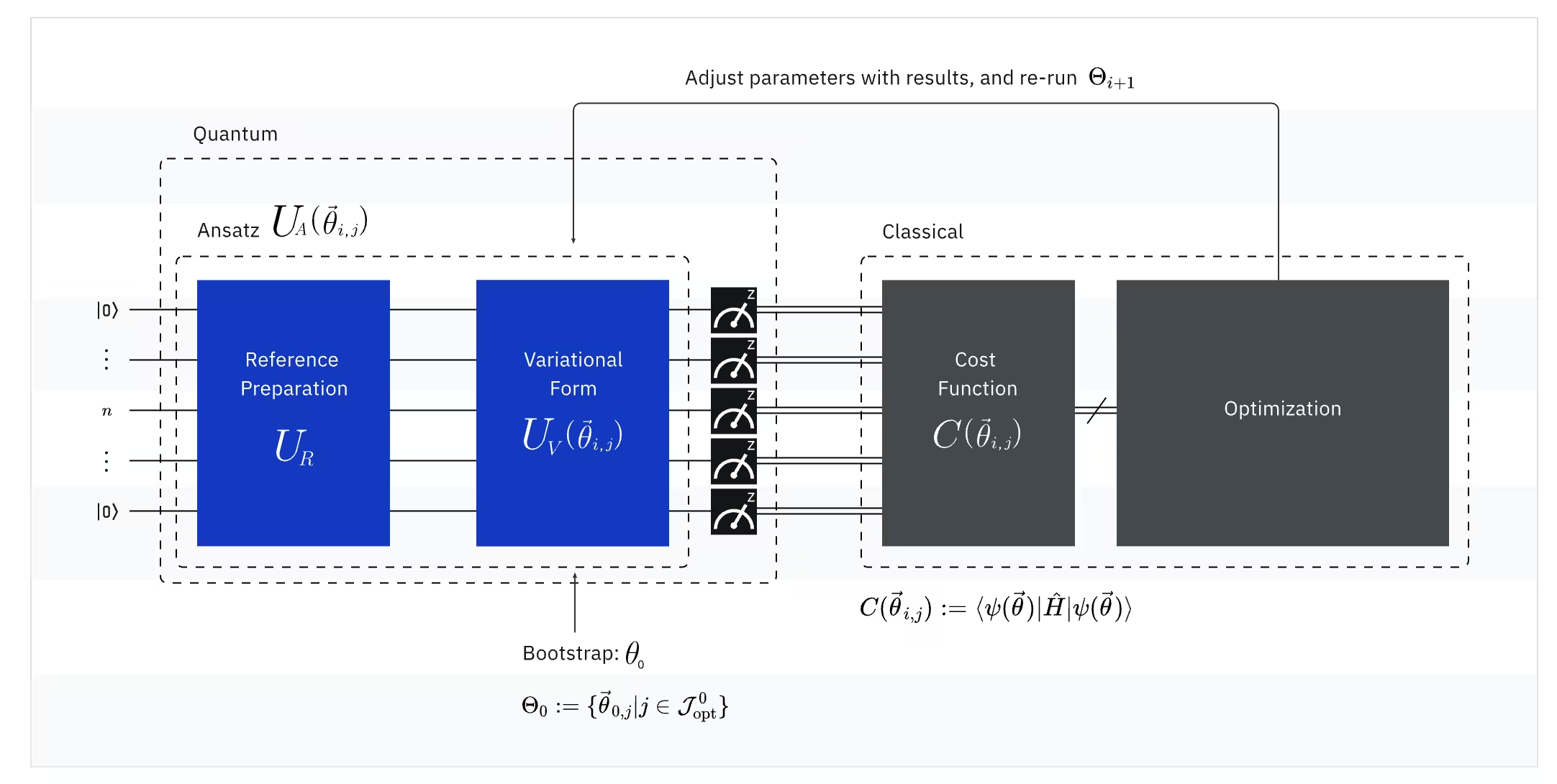
(Kuantum) Observable: Hamiltonian molekul (energi sebuah molekul)
Dalam VQE, Hamiltonian molekuler/atom adalah sebuah observable, yang berarti kita dapat mengukur nilainya melalui sebuah eksperimen. Tujuan kita adalah menemukan energi serendah mungkin (energi ground state) dari molekul. Untuk melakukan ini, kita menggunakan keadaan kuantum trial, yang dihasilkan oleh Circuit kuantum yang diparameterisasi (ansatz). Kita mengukur observable dan mengoptimalkan keadaan kuantum hingga kita mencapai energi serendah mungkin.
Basis set yang digunakan untuk Hamiltonian molekul menentukan jumlah qubit yang dibutuhkan dan secara langsung mempengaruhi akurasi VQE. Memilih basis set yang tepat sangat penting untuk menyeimbangkan efisiensi dan presisi. Untuk menyederhanakan perhitungan tanpa mengubah basis set, kita dapat menggunakan strategi seperti pemberlakuan simetri dan pengurangan ruang aktif. Banyak molekul memiliki bentuk simetris (seperti kupu-kupu atau kepingan salju), artinya beberapa bagian berperilaku dengan cara yang sama. Alih-alih menghitung semuanya secara terpisah, kita dapat fokus hanya pada bagian yang unik, menghemat sumber daya kuantum, sehingga memanfaatkan simetri. Dalam pengurangan ruang aktif, kita hanya mempertimbangkan orbital yang penting, karena tidak semua elektron secara signifikan mempengaruhi energi molekul. Elektron yang dekat dengan inti sebagian besar tetap tidak berubah, sementara yang lain mempengaruhi ikatan. Dengan menerapkan metode-metode ini, kita dapat membuat VQE lebih efisien sambil mempertahankan akurasi.
Setelah kita mendapatkan Hamiltonian molekul menggunakan basis set yang tepat dan strategi di atas, kita perlu mengubah Hamiltonian ini menjadi yang cocok untuk komputer kuantum. Memetakan masalah ke operator Pauli bisa sangat rumit. Ini terutama berlaku dalam kimia kuantum, yang bekerja dengan partikel yang tidak dapat dibedakan (elektron), karena qubit dapat dibedakan. Kita tidak akan membahas detail pemetaan di sini, tetapi kami merujuk kamu ke sumber daya berikut. Diskusi umum tentang pemetaan masalah ke operator kuantum dapat ditemukan di Quantum computing in practice. Diskusi lebih rinci tentang pemetaan masalah kimia ke operator kuantum dapat ditemukan di Quantum chemistry with VQE.
Untuk modul ini, kami akan menyediakan Hamiltonian (satu qubit) yang sesuai untuk dan sehingga kita dapat fokus menggunakan komputer kuantum. Hamiltonian satu qubit ini disiapkan menggunakan basis set STO-6G dan pemetaan Jordan-Wigner, yang merupakan pemetaan paling langsung dengan interpretasi fisik paling sederhana, karena ia memetakan hunian satu spin-orbital ke hunian satu qubit. Selain itu, kami menggunakan teknik pengurangan qubit dengan menggunakan simetri Hamiltonian, yang menggunakan pola dalam bagaimana hunian spin berperilaku untuk mengurangi jumlah qubit. Untuk molekul , kita mengasumsikan jarak antara dua atom hidrogen adalah 0.735 .
(Kuantum) Ansatz: Fungsi gelombang trial (Bagaimana membangun keadaan kuantum trial dengan Circuit kuantum)
Untuk VQE, ansatz (jamak: ansätze) terdiri dari dua komponen utama. Yang pertama adalah persiapan keadaan awal, yang menyiapkan keadaan qubit dengan menerapkan Gate kuantum tanpa parameter variasional. Komponen kedua adalah Circuit kuantum yang diparameterisasi, sebuah sirkuit kuantum khusus dengan parameter yang dapat disesuaikan, mirip dengan kenop di radio. Parameter-parameter ini akan digunakan untuk bagian terakhir — optimizer klasik — untuk membantu kita mencapai ground state terbaik yang mungkin.
Dalam bagian prinsip variasional, kita mempelajari bahwa kualitas keadaan trial mempengaruhi kualitas hasil algoritma variasional. Ini berarti bahwa memilih ansatz yang baik sangat penting dalam VQE. Sekali lagi, ini adalah topik yang kaya dan kompleks. Kita tidak akan membahas berbagai jenis ansatz atau asal-usulnya di sini. Jika kamu tertarik mempelajari lebih lanjut tentang Circuit kuantum yang diparameterisasi dan ansatz, kamu dapat menjelajahi pelajaran Ansatz and variational form dari kursus Variational algorithm design, yang menyediakan penjelasan dan contoh ansätze secara rinci.
Karena kita akan menggunakan Hamiltonian satu qubit dalam modul ini, kita memerlukan Circuit kuantum yang diparameterisasi satu qubit sebagai ansatz. Kita akan melihat tiga jenis ansätze satu qubit pada bagian berikut. Kita akan membandingkannya dan mendiskusikan pertimbangan utama dalam memilih ansatz.
(Klasik) Optimizer: menyempurnakan Circuit kuantum
Setelah komputer kuantum mengukur energi observable dari ansatz, parameter ansatz dan nilai energi dikirim ke optimizer klasik untuk disempurnakan. Proses optimasi ini dilakukan pada komputer klasik, biasanya menggunakan paket ilmiah serba guna seperti SciPy.
Optimizer klasik memperlakukan energi yang diukur sebagai fungsi biaya. Dalam masalah optimasi, fungsi biaya (kadang juga disebut fungsi objektif) adalah fungsi matematis yang mengukur seberapa "baik" solusi tertentu. Tujuan optimizer adalah menemukan set parameter yang meminimalkan fungsi biaya ini. Dalam konteks menemukan energi ground state molekul, energi itu sendiri berfungsi sebagai fungsi biaya — kita ingin menemukan parameter untuk Circuit kuantum kita (sebuah "solusi") yang menghasilkan energi serendah mungkin. Optimizer klasik menggunakan nilai energi yang diukur ini (biaya) dan menentukan set parameter yang dioptimalkan berikutnya untuk ansatz kuantum. Parameter yang diperbarui ini kemudian dikirim kembali ke Circuit kuantum, dan proses diulang. Dengan setiap iterasi, optimizer klasik menyesuaikan parameter untuk mencoba mengurangi energi (meminimalkan fungsi biaya) hingga kriteria konvergensi yang telah ditentukan terpenuhi, idealnya memastikan bahwa energi serendah mungkin (sesuai dengan ground state molekul untuk jarak ikatan dan basis set tersebut) ditemukan.
Ada banyak strategi optimasi yang disediakan oleh paket ilmiah seperti SciPy. Kamu dapat menemukan lebih banyak di pelajaran Optimization loops dari kursus Variational algorithm design. Di sini kita akan menggunakan COBYLA (Constrained Optimization BY Linear Approximations), sebuah algoritma optimasi yang cocok untuk lanskap energi yang rumit. Secara khusus, COBYLA tidak mencoba menghitung gradien dari fungsi yang dipelajari; ini disebut optimizer bebas gradien. Bayangkan kamu sedang mencoba menemukan puncak tertinggi di pegunungan dengan mata tertutup. Karena kamu tidak bisa melihat seluruh lanskap, kamu mengambil langkah kecil ke berbagai arah, sambil memeriksa apakah kamu naik atau turun. COBYLA bekerja dengan cara serupa — ia bergerak melalui ruang parameter, menguji nilai-nilai yang berbeda, secara bertahap meningkatkan hasil hingga menemukan yang terbaik.
Sekarang kamu siap untuk melakukan perhitungan VQE. Untuk itu, coba pertanyaan check-in di bawah ini, yang merangkum keseluruhan proses.
Cek pemahamanmu
Isi titik-titik dengan istilah yang tepat untuk melengkapi ringkasan proses VQE, lalu klik untuk memeriksa jawabanmu.
VQE adalah algoritma kuantum variasional, yang menggabungkan kekuatan (1) ________ dan komputasi klasik, yang digunakan untuk menemukan (2) __________ sebuah molekul. Proses dimulai dengan mendefinisikan (3) __________, yang mewakili total energi sistem dan bertindak sebagai observable dalam pengukuran kuantum. Selanjutnya, kita menyiapkan sebuah (4) __________, sebuah sirkuit kuantum dengan parameter yang dapat disesuaikan yang mewakili fungsi gelombang trial molekul. Parameter-parameter ini dioptimalkan menggunakan (5) __________, sebuah algoritma klasik yang menyesuaikan parameter secara iteratif untuk meminimalkan energi yang diukur. Dalam diskusi di atas kita menggunakan optimizer (6) __________, yang menyempurnakan parameter ansatz tanpa memerlukan perhitungan turunan. Proses berlanjut hingga kita mencapai (7) __________, artinya kita telah menemukan energi terendah yang mungkin dari molekul.
Word Bank:
- classical optimizer
- ground state energy
- hardware-efficient
- ansatz
- molecular Hamiltonian
- COBYLA
- quantum computing
- convergence
Jawaban
1 → quantum computing
2 → ground state energy
3 → molecular Hamiltonian
4 → ansatz
5 → classical optimizer
6 → COBYLA
7 → convergence
Menghitung energi ground state atom hidrogen dengan VQE
Sekarang, mari kita gunakan apa yang sudah kita pelajari untuk menghitung energi ground state atom hidrogen. Sepanjang modul ini, kita akan menggunakan kerangka kerja komputasi kuantum yang dikenal sebagai "Qiskit patterns", yang membagi alur kerja menjadi langkah-langkah berikut:
- Langkah 1: Petakan input klasik ke masalah kuantum
- Langkah 2: Optimalkan masalah untuk eksekusi kuantum
- Langkah 3: Eksekusi menggunakan Qiskit Runtime primitives
- Langkah 4: Post-processing dan analisis klasik

Kita akan mengikuti langkah-langkah ini secara umum.
Mari mulai dengan memuat beberapa paket yang diperlukan, termasuk Qiskit Runtime primitives. Kita juga akan memilih komputer kuantum yang paling tidak sibuk yang tersedia.
Ada kode di bawah untuk menyimpan kredensial kamu saat pertama kali digunakan. Pastikan untuk menghapus informasi ini dari notebook setelah menyimpannya ke lingkunganmu, agar kredensial tidak terbagi secara tidak sengaja saat kamu berbagi notebook. Lihat Set up your IBM Cloud account dan Initialize the service in an untrusted environment untuk panduan lebih lanjut.
# Load the Qiskit Runtime service
from qiskit_ibm_runtime import QiskitRuntimeService
# Load the Runtime primitive and session
from qiskit_ibm_runtime import EstimatorV2 as Estimator
# Syntax for first saving your token. Delete these lines after saving your credentials.
# QiskitRuntimeService.save_account(channel='ibm_quantum_platform',
# instance = '<YOUR_IBM_INSTANCE_CRN>', token='<YOUR-API_KEY>', overwrite=True, set_as_default=True)
# service = QiskitRuntimeService(channel='ibm_quantum_platform')
# Load saved credentials
service = QiskitRuntimeService()
# Use the least busy backend, or uncomment the loading of a specific backend like "ibm_brisbane".
backend = service.least_busy(operational=True, simulator=False, min_num_qubits=127)
# backend = service.backend("ibm_brisbane")
print(backend.name)
ibm_brisbane
Sel di bawah ini memungkinkan kamu untuk beralih antara menggunakan simulator atau hardware nyata sepanjang notebook. Kami rekomendasikan untuk menjalankannya sekarang:
# Load the Aer simulator and generate a noise model based on the currently-selected backend.
from qiskit_aer import AerSimulator
from qiskit_aer.noise import NoiseModel
# Alternatively, load a fake backend with generic properties and define a simulator.
noise_model = NoiseModel.from_backend(backend)
# Define a simulator using Aer, and use it in Sampler.
backend_sim = AerSimulator(noise_model=noise_model)
Langkah 1: Petakan masalah ke Circuit dan operator kuantum
Kita mulai perhitungan VQE dengan mendefinisikan Hamiltonian untuk molekul hidrogen () pada jarak ikatan tertentu. Hamiltonian ini mewakili total energi sistem dalam bentuk operator qubit, yang telah diproduksi dan dipetakan dari sistem molekul menggunakan prosedur standar: 1) menggunakan basis set STO-6G (kumpulan fungsi matematika tertentu yang digunakan untuk mengaproksimasi orbital elektron), 2) menerapkan pemetaan Jordan-Wigner (teknik untuk menerjemahkan operator fermionik yang menggambarkan elektron ke dalam operator qubit), dan 3) melakukan reduksi qubit menggunakan paritas Hamiltonian untuk menyederhanakan masalah.
Seperti yang sudah kita jelaskan sebelumnya, energi ground state yang dihitung sangat bergantung pada pemilihan basis set dan geometri molekul (seperti jarak ikatan). Untuk konfigurasi spesifik ini dan setelah transformasi-transformasi ini, Hamiltonian qubit yang dihasilkan cukup sederhana:
Di sini, mewakili operator identitas dan mewakili operator Pauli-Z, yang bekerja pada satu qubit. Koefisien-koefisiennya diturunkan dari integral yang dihitung menggunakan basis set STO-6G pada jarak ikatan tertentu ini dengan transformasi yang tepat.
Dengan Hamiltonian ini terdefinisi, kita sekarang dapat menggunakan VQE untuk menghitung energi ground state-nya. Ada baiknya membandingkan energi ground state yang kita hitung dengan nilai yang diharapkan. Untuk atom hidrogen tunggal yang terisolasi (H), energi ground state-nya tepat -0,5 Hartree (tanpa adanya efek relativistik). Mari kita hitung energi ground state yang tepat dari Hamiltonian qubit spesifik kita seperti yang didefinisikan di atas dan bandingkan dengan nilai-nilai yang diketahui secara relevan.
from qiskit.quantum_info import SparsePauliOp
import numpy as np
# Qubit Hamiltonian of the hydrogen atom generated by using STO-3G basis set and parity mapping
Hamiltonian = SparsePauliOp.from_list([("I", -0.2355), ("Z", 0.2355)])
# exact ground state energy of Hamiltonian
A = np.array(Hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print(
"The exact ground state energy of the Hamiltonian is ",
min(eigenvalues).real,
"hartree",
)
h = min(eigenvalues.real)
The exact ground state energy of the Hamiltonian is -0.471 hartree
Selanjutnya, kita membutuhkan Circuit kuantum yang diparameterisasi, sebuah ansatz, untuk menyiapkan fungsi gelombang percobaan untuk ground state. Tujuannya adalah menemukan parameter yang meminimalkan nilai ekspektasi energi . Pemilihan ansatz sangat krusial karena menentukan sekumpulan kemungkinan state kuantum yang dapat disiapkan oleh Circuit kita. Ansatz yang "baik" adalah yang cukup fleksibel untuk merepresentasikan state yang sangat mendekati ground state sebenarnya dari Hamiltonian yang kita pelajari, namun tidak terlalu kompleks sehingga membutuhkan terlalu banyak parameter atau Circuit yang terlalu dalam untuk komputer kuantum saat ini.
Di sini, kita akan mencoba tiga ansatz satu-qubit yang berbeda untuk melihat mana yang memberikan "jangkauan" yang lebih baik atas kemungkinan state kuantum yang dapat dimiliki satu qubit. "Jangkauan" di sini mengacu pada rangkaian state kuantum yang dapat diproduksi oleh Circuit ansatz dengan memvariasikan parameternya.
Kita akan menggunakan tiga ansatz berdasarkan kombinasi berbeda dari Gate rotasi satu-qubit:
- Satu ansatz Gate rotasi 1-sumbu: Ansatz ini menggunakan rotasi hanya di sekitar satu sumbu (). Pada Bloch sphere, ini bersesuaian dengan bergerak hanya di sepanjang lingkaran tertentu. Ini yang paling tidak fleksibel dan hanya mencakup sekumpulan state yang terbatas.
- Dua ansatz Gate rotasi 2-sumbu: Ansatz-ansatz ini menggabungkan rotasi di sekitar dua sumbu yang berbeda ( dan ). Ini memungkinkan kita menjangkau bagian yang lebih besar dari Bloch sphere, dibandingkan dengan rotasi satu sumbu.
Dengan membandingkan hasil VQE yang diperoleh dengan ketiga ansatz ini, kita dapat melihat bagaimana fleksibilitas dan jangkauan state-space dari ansatz memengaruhi kemampuan kita untuk menemukan energi ground state sebenarnya dari Hamiltonian sederhana kita. Ansatz yang lebih fleksibel memiliki potensi untuk menemukan aproksimasi yang lebih baik, tetapi mungkin juga lebih sulit bagi optimizer klasik.
from qiskit import QuantumCircuit
from qiskit.circuit import Parameter
from qiskit.quantum_info import Statevector, DensityMatrix, Pauli
theta = Parameter("θ")
phi = Parameter("φ")
lam = Parameter("λ")
ansatz1 = QuantumCircuit(1)
ansatz1.rx(theta, 0)
ansatz2 = QuantumCircuit(1)
ansatz2.rx(theta, 0)
ansatz2.rz(phi, 0)
ansatz3 = QuantumCircuit(1)
ansatz3.rx(theta, 0)
ansatz3.rz(phi, 0)
ansatz3.rx(lam, 0)
<qiskit.circuit.instructionset.InstructionSet at 0x1059def80>
Sekarang, mari kita hasilkan 5000 bilangan acak untuk setiap parameter dan plot distribusi state kuantum acak yang dihasilkan oleh ketiga ansatz dengan parameter acak ini. Kamu bisa menganggap parameter-parameter ini seperti rotasi di sekitar sumbu-sumbu yang berbeda pada permukaan bola. Untuk melihat distribusi state kuantum, kita akan menggunakan Bloch Sphere, bola tiga dimensi yang menunjukkan state satu qubit. Setiap titik pada bola mewakili kemungkinan state qubit, di mana kutub utara dan selatan seperti "0" dan "1" klasik, tetapi qubit juga bisa berada di antara keduanya, menunjukkan sifat kuantum khusus seperti superposisi. Pertama, siapkan fungsi-fungsi yang diperlukan untuk memplot Bloch sphere 3D dan siapkan 5000 parameter acak.
import matplotlib.pyplot as plt
def plot_bloch(bloch_vectors):
# Extract X, Y, Z coordinates for 3D projection
X_coords = bloch_vectors[:, 0]
Z_coords = bloch_vectors[:, 2]
# Compute Y coordinates from X and Z to approximate the full Bloch sphere projection
Y_coords = bloch_vectors[:, 1]
# Create 3D plot
fig = plt.figure(figsize=(8, 8))
ax = fig.add_subplot(111, projection="3d")
ax.scatter(X_coords, Y_coords, Z_coords, color="blue", alpha=0.6)
# Labels and title
ax.set_xlabel("X")
ax.set_ylabel("Y")
ax.set_zlabel("Z")
ax.set_title("Parameterized 1-Qubit Circuit on 3D Bloch Sphere")
# Set axis limits and make them equal
ax.set_xlim([-1, 1])
ax.set_ylim([-1, 1])
ax.set_zlim([-1, 1])
# Ensure equal aspect ratio for all axes
ax.set_box_aspect([1, 1, 1]) # Equal scaling for x, y, z axes
# Show grid
ax.grid(True)
plt.show()
num_samples = 5000 # Number of random states
theta_vals = np.random.uniform(0, 2 * np.pi, num_samples)
phi_vals = np.random.uniform(0, 2 * np.pi, num_samples)
lam_vals = np.random.uniform(0, 2 * np.pi, num_samples)
Mari lihat bagaimana ansatz pertama kita bekerja.
# List to store Bloch Sphere XZ coordinates
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create a circuit and bind parameters
qc = ansatz1
bound_qc = qc.assign_parameters({theta: theta_vals[i]}) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to a numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
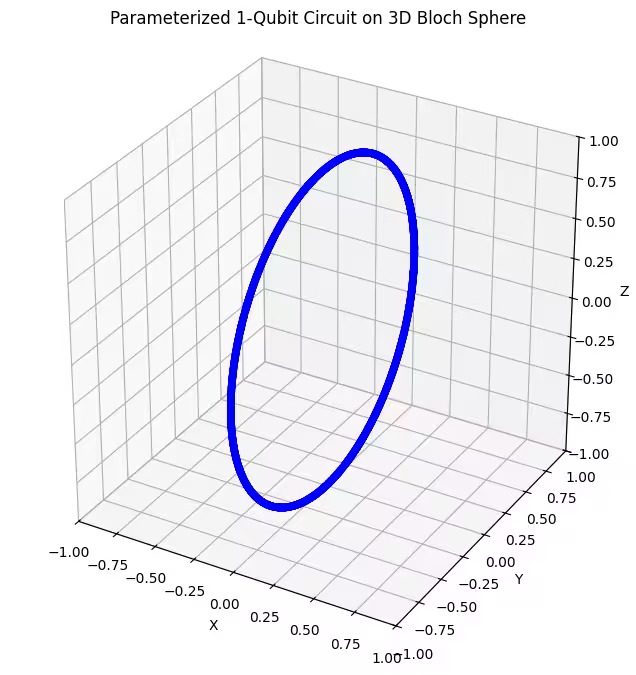
Kita bisa lihat bahwa ansatz pertama kita menghasilkan state kuantum berbentuk cincin yang terdistribusi pada Bloch sphere. Ini masuk akal, karena kita hanya memberikan satu parameter rotasi pada ansatz. Oleh karena itu, ia hanya bisa menghasilkan state yang dirotasikan di sekitar satu sumbu. Mulai dari titik dan memutar di sekitar satu sumbu akan selalu menghasilkan sebuah cincin. Lalu mari kita periksa ansatz kedua kita, yang memiliki dua Gate rotasi ortogonal - Rx dan Rz.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz2
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i]}
) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
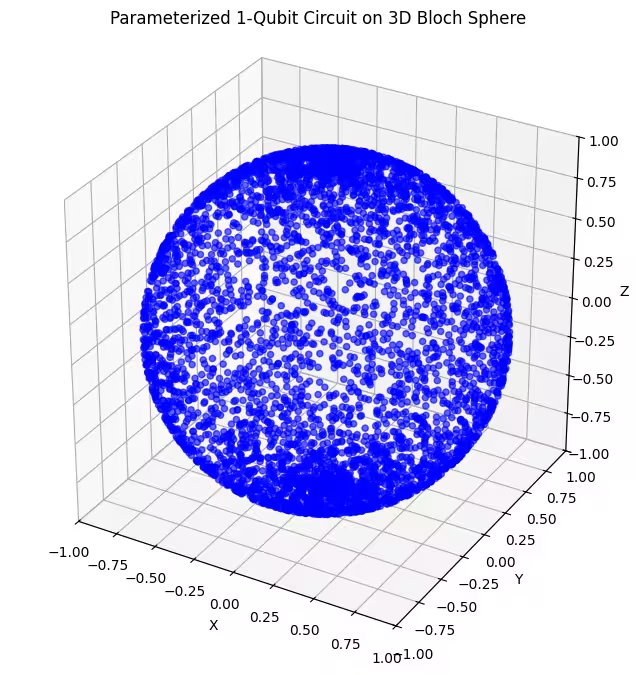
Di sini, kita bisa lihat bahwa ansatz kedua kita mencakup bagian yang lebih besar dari Bloch sphere - tetapi perhatikan bahwa titik-titiknya lebih terkonsentrasi di sekitar kutub dan lebih tersebar di sekitar ekuator. Sekarang saatnya memeriksa ansatz terakhir kita.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz3
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i], lam: lam_vals[i]}
)
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
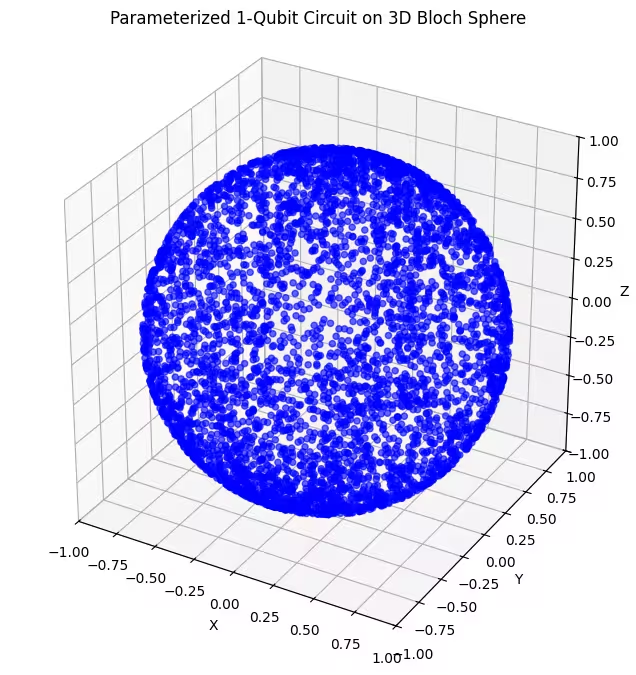
Di sini kamu bisa lihat state kuantum yang lebih merata terdistribusi yang dihasilkan oleh ansatz terakhir kita.
Seperti yang disebutkan, hal terbaik yang bisa dilakukan adalah mendapatkan pengetahuan tentang ground state yang kamu cari dan menggunakan ansatz yang cocok untuk menjangkau state yang mendekati ground state tersebut. Misalnya, jika kita tahu bahwa ground state kita ada di dekat kutub, kita mungkin akan memilih ansatz 2. Demi kesederhanaan, kita akan menggunakan ansatz 3, yang secara merata menjangkau seluruh Bloch sphere.
Sekarang setelah kita memilih ansatz kita, mari kita gambar Circuit-nya.
# Pre-defined ansatz circuit and operator class for Hamiltonian
ansatz = ansatz3
num_params = ansatz.num_parameters
print("This circuit has ", num_params, "parameters")
ansatz.draw("mpl", style="iqp")
This circuit has 3 parameters

Langkah 2: Optimalkan untuk hardware target
Saat menjalankan perhitungan pada komputer kuantum nyata, kita tidak hanya peduli tentang logika Circuit kuantum. Kita juga peduli tentang hal-hal seperti operasi apa yang bisa dilakukan oleh komputer kuantum tertentu tersebut, dan di mana qubit yang kita gunakan berada pada komputer kuantum tersebut. Apakah mereka berdekatan satu sama lain? Apakah mereka berjauhan? Oleh karena itu, langkah berikutnya adalah menulis ulang Circuit kita menggunakan Gate yang natural untuk komputer kuantum yang akan kita gunakan, dan mempertimbangkan tata letak qubit. Ini bisa dilakukan dengan transpilation - setelah proses ini, kamu bisa lihat ansatz sederhana kita diubah menjadi sekumpulan Gate yang berbeda, dan qubit abstrak kita akan dipetakan ke qubit fisik pada komputer kuantum nyata.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
config = backend.configuration()
print("Backend: {config.backend_name}")
print("Native gates: ", config.supported_instructions, ",")
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa = pm.run(ansatz)
ansatz_isa.draw(output="mpl", idle_wires=False, style="iqp")
Backend: {config.backend_name}
Native gates: ['ecr', 'id', 'delay', 'measure', 'reset', 'rz', 'sx', 'x'] ,

Kamu bisa lihat Gate rx, rz dari ansatz kita diubah menjadi serangkaian Gate rz, sx, yang merupakan Gate native dari Backend kita. Juga, kamu bisa lihat q0 kita sekarang dipetakan ke qubit fisik kelima. Kita juga perlu memetakan Hamiltonian kita sesuai dengan perubahan-perubahan ini, seperti pada kode berikut:
Hamiltonian_isa = Hamiltonian.apply_layout(layout=ansatz_isa.layout)
Langkah 3: Eksekusi pada hardware target
Sekarang saatnya menjalankan VQE kita pada QPU nyata. Untuk ini, pertama kita membutuhkan fungsi biaya untuk proses optimasi, yang mengevaluasi nilai ekspektasi Hamiltonian dengan state kuantum yang dihasilkan oleh ansatz. Jangan khawatir! Kamu tidak perlu membuat kode semuanya sendiri. Kami sudah menyiapkan fungsi untuk ini, dan yang perlu kamu lakukan hanyalah menjalankan sel di bawah ini.
def cost_func(params, ansatz, hamiltonian, estimator):
"""Return estimate of energy from estimator
Parameters:
params (ndarray): Array of ansatz parameters
ansatz (QuantumCircuit): Parameterized ansatz circuit
hamiltonian (SparsePauliOp): Operator representation of Hamiltonian
estimator (EstimatorV2): Estimator primitive instance
cost_history_dict: Dictionary for storing intermediate results
Returns:
float: Energy estimate
"""
pub = (ansatz, [hamiltonian], [params])
result = estimator.run(pubs=[pub]).result()
energy = result[0].data.evs[0]
cost_history_dict["iters"] += 1
cost_history_dict["prev_vector"] = params
cost_history_dict["cost_history"].append(energy)
print(f"Iters. done: {cost_history_dict['iters']} [Current cost: {energy}]")
return energy
Akhirnya, kita siapkan parameter awal untuk ansatz kita dan proses optimasinya. Kamu bisa menggunakan semua nol atau nilai acak. Kita sudah memilih parameter awal di bawah ini, tetapi jangan ragu untuk memberi komentar atau menghapus komentar pada baris di sel untuk mengambil sampel parameter secara acak, seragam dari 0 hingga .
# x0 = np.random.uniform(0, 2*pi, 3)
x0 = [1, 1, 0]
# QPU Est. 2min for ibm_brisbane
from scipy.optimize import minimize
from qiskit_ibm_runtime import Batch
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, Hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 10, "tol": 0.01},
)
batch.close()
Iters. done: 1 [Current cost: -0.3361517318448143]
Iters. done: 2 [Current cost: -0.4682546422099432]
Iters. done: 3 [Current cost: -0.38985802144149584]
Iters. done: 4 [Current cost: -0.38319217316749354]
Iters. done: 5 [Current cost: -0.4628720756579032]
Iters. done: 6 [Current cost: -0.4683301936226905]
Iters. done: 7 [Current cost: -0.45480498699294747]
Iters. done: 8 [Current cost: -0.4690533242050814]
Iters. done: 9 [Current cost: -0.465867415110354]
Iters. done: 10 [Current cost: -0.4606882723137227]
h_vqe = res.fun
print("The reference ground state energy is ", min(eigenvalues))
print("The computed ground state energy is ", h_vqe)
The reference ground state energy is (-0.471+0j)
The computed ground state energy is -0.4690533242050814
Selamat! Kamu baru saja berhasil menyelesaikan eksperimen kimia kuantum pertamamu. Kita bisa melihat perbedaan antara energi ground state Hamiltonian yang tepat dan milik kita, tetapi karena kita menggunakan teknik mitigasi error default (yang mengoreksi error readout), perbedaannya kecil. Ini adalah awal yang sangat baik!
Catatan: Kamu bisa mendapatkan hasil yang lebih baik dengan menetapkan tingkat mitigasi error menggunakan resilience_level. Nilai defaultnya adalah 1, dan jika kamu menetapkan nilai yang lebih tinggi, ini akan menggunakan lebih banyak waktu QPU tetapi mungkin menghasilkan hasil yang lebih baik.
Langkah 4: Post-process
Saatnya melihat bagaimana optimizer klasik kita bekerja. Jalankan sel di bawah ini dan lihat pola konvergensinya.
fig, ax = plt.subplots()
x = np.linspace(0, 10, 10)
# Define the constant function
y_constant = np.full_like(x, h)
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

Kita mulai dengan nilai awal yang cukup baik, sehingga kita mendapatkan nilai akhir yang baik hanya dalam 10 langkah. Kamu bisa melihat puncak besar dan kecil, dan ini adalah fitur khas dari optimizer COBYLA - ia menjelajahi ruang seolah-olah tidak bisa melihat lanskap dan menyesuaikan ukuran langkah dengan setiap pengukuran.
Cek pemahamanmu
Apa pengamatanmu? Bagian mana dari proses di atas yang terbuka untuk perbaikan guna mendapatkan hasil yang lebih mendekati nilai teoritis, atau lebih mendekati energi ground state yang tepat dari Hamiltonian? Apa saja hal yang perlu dipertimbangkan untuk ini?
Jawaban
Hal pertama yang perlu dipertimbangkan adalah perubahan dalam kumpulan basis yang digunakan dalam menghitung Hamiltonian molekul. Seperti yang disebutkan sebelumnya, energi ground state atom H adalah -0,5 Hartree, seperti yang sudah diketahui dengan baik, dan basis STO-6G yang kita pilih tidak cukup untuk menurunkan nilai ini secara akurat.
Memilih jenis basis yang lebih kompleks meningkatkan jumlah qubit yang digunakan oleh Hamiltonian; oleh karena itu, kita perlu memilih ansatz yang lebih kompleks dan cocok untuk masalah kimia.
Selanjutnya yang perlu dioptimalkan adalah pengelolaan noise dalam QPU. Teknik mitigasi error yang lebih canggih menghasilkan hasil yang lebih baik tetapi mungkin membutuhkan waktu lebih lama untuk digunakan. Juga, pertimbangkan bagaimana shot_number memengaruhi hasilnya.
Terakhir, kinerja konvergensi yang lebih baik juga bisa dicapai dengan mencoba optimizer yang berbeda.
Menghitung energi ground state molekul hidrogen dengan VQE
Sekarang setelah kita melihat keseluruhan proses VQE menggunakan atom , kita akan menghitung energi ground state molekul dengan lebih cepat.
Langkah 1: Petakan masalah ke Circuit dan operator kuantum
Di sini kita juga menyediakan Hamiltonian satu-qubit yang menggunakan basis STO-6G dan transformasi Jordan-Wigner, dengan reduksi qubit menggunakan simetri Hamiltonian. Perhatikan bahwa kita menggunakan jarak atom antara dua atom hidrogen sebesar 0.735 .
Tidak seperti perhitungan atom hidrogen tunggal (), untuk menghitung ground state molekul hidrogen (), kita juga harus mempertimbangkan gaya tolak yang bekerja antara nuklei dua atom hidrogen, selain energi yang terkait dengan orbital elektronik. Pada langkah ini, kita akan memberikan nilai ini sebagai konstanta, dan kita akan benar-benar menghitung nilai ini dalam soal check-in.
h2_hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
# exact ground state energy of hamiltonian
nuclear_repulsion = 0.71997
A = np.array(h2_hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print("Electronic ground state energy (Hartree): ", min(eigenvalues).real)
print("Nuclear repulsion energy (Hartree): ", nuclear_repulsion)
print(
"Total ground state energy (Hartree): ", min(eigenvalues).real + nuclear_repulsion
)
h2 = min(eigenvalues).real + nuclear_repulsion
Electronic ground state energy (Hartree): -1.8659468547627318
Nuclear repulsion energy (Hartree): 0.71997
Total ground state energy (Hartree): -1.1459768547627318
Langkah 2: Optimalkan untuk hardware target
Karena jumlah qubit yang digunakan oleh VQE dan Hamiltonian sebelumnya sama dengan Backend yang akan digunakan untuk eksekusi, kita akan menggunakan ansatz yang sudah ada beserta bentuk yang sudah dioptimalkan.
h2_hamiltonian_isa = h2_hamiltonian.apply_layout(layout=ansatz_isa.layout)
Langkah 3: Eksekusi pada hardware target
Sekarang saatnya melakukan perhitungan pada QPU yang sebenarnya. Hampir semuanya sama, tetapi kita akan menggunakan titik awal yang sesuai untuk menyesuaikan Hamiltonian. Juga, pada bagian iteratif, beberapa pengaturan Estimator, yang digunakan untuk menghitung ekspektasi Hamiltonian untuk ansatz di QPU, akan diatur sedikit berbeda dari perhitungan sebelumnya. Kita akan membahas perubahan ini lebih lanjut dalam pertanyaan check-in.
x0 = [2, 0, 0]
# QPU time 4min for ibm_brisbane
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, h2_hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 15},
)
batch.close()
Iters. done: 1 [Current cost: -0.710621837568328]
Iters. done: 2 [Current cost: -0.2603208441168329]
Iters. done: 3 [Current cost: -0.25548711201326424]
Iters. done: 4 [Current cost: -0.581129450619904]
Iters. done: 5 [Current cost: -1.722920997605439]
Iters. done: 6 [Current cost: -1.6633324849371915]
Iters. done: 7 [Current cost: -1.8066989598929164]
Iters. done: 8 [Current cost: -1.8051093803839542]
Iters. done: 9 [Current cost: -1.802692217571555]
Iters. done: 10 [Current cost: -1.8233585485263144]
Iters. done: 11 [Current cost: -1.6904116652617205]
Iters. done: 12 [Current cost: -1.8245120321245392]
Iters. done: 13 [Current cost: -1.6837021361383608]
Iters. done: 14 [Current cost: -1.8166632606115467]
Iters. done: 15 [Current cost: -1.863446212658907]
h2_vqe = res.fun + nuclear_repulsion
print(
"The reference ground state energy is ", min(eigenvalues).real + nuclear_repulsion
)
print("The computed ground state energy is ", h2_vqe)
The reference ground state energy is -1.1459768547627318
The computed ground state energy is -1.143476212658907
Meskipun VQE secara teoritis memberikan batas atas terhadap energi ground state yang sebenarnya, implementasi praktis pada hardware kuantum nyata atau berderau yang disimulasikan, serta aproksimasi yang dibuat dalam menyiapkan Hamiltonian (seperti basis set atau reduksi qubit), dapat menimbulkan error yang terkadang menghasilkan energi yang diukur sedikit lebih rendah dari nilai teoritis yang tepat atau referensi numerik tertentu. Meskipun ada beberapa error, hasilnya tampak memuaskan, terutama mengingat jumlah langkah yang kecil. Sekarang, mari selesaikan perhitungan VQE ini dengan melihat bagaimana optimizer bekerja.
Langkah 4: Post-process
fig, ax = plt.subplots()
x = np.linspace(0, 5, 15)
# Define the constant function
y_constant = np.full_like(x, min(eigenvalues))
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

Cek pemahamanmu
Mari hitung energi tolak nuklir molekul , yang kita sertakan sebagai nilai konstanta (0,71997 Hartree).

Gunakan hukum Coulomb dan satuan atom untuk memastikan kamu mendapatkan nilai dalam Hartree.
Jawaban
Karena kedua nuklei hidrogen bermuatan positif, mereka saling tolak karena gaya elektrostatik. Tolakan ini digambarkan oleh hukum Coulomb:
di mana adalah muatan proton, adalah permitivitas vakum, dan adalah jarak antara dua nuklei, diukur dalam meter atau jari-jari Bohr dalam satuan joule (J).
Untuk menghitung energi ini dalam Hartree, kita perlu mengubah persamaan di atas ke dalam sistem Satuan Atom (AU). Dalam AU, , dan jari-jari Bohr () adalah 1 dan menjadi skala panjang fundamental dalam AU. Dengan penyederhanaan ini, hukum Coulomb menjadi:
di mana harus diukur dalam jari-jari Bohr ().
Untuk mengubah pemisahan nuklir yang diberikan dalam ke , kita membutuhkan relasi konversi ini:
sehingga menjadi .
Oleh karena itu, energi tolak nuklir dari yang diberikan adalah
Hitung energi reaksi
Sekarang mari kita gunakan apa yang sudah kita dapatkan! Kamu sudah menggunakan VQE, variational quantum eigensolver, untuk menghitung energi ground state dari atom dan molekul . Yang tersisa adalah menggunakan nilai-nilai yang sudah dihitung untuk mendapatkan energi reaksi dari proses .
Energi reaksi adalah perubahan energi yang terjadi ketika zat-zat bereaksi membentuk zat baru. Bayangkan kamu sedang membangun sesuatu: kadang kamu perlu memasukkan energi ke dalamnya (seperti menumpuk balok), dan kadang energi dilepaskan (seperti bola menggelinding ke bawah bukit). Dalam kimia, reaksi bisa menyerap energi (endotermik) atau melepaskan energi (eksotermik).
Energi reaksi dari proses dapat dihitung dengan rumus berikut:
Dengan menjalankan sel di bawah ini, mari kita lihat secara visual. Di sini kita akan menggunakan nilai ground state eksak dari setiap Hamiltonian, dan kita akan membandingkan energi reaksi dari solusi eksak dan hasil VQE.
# Theoretical values
E_H_theo = h.real
E_H2_theo = h2
# Experimental values
E_H_exp = h_vqe
E_H2_exp = h2_vqe
# Calculate reaction energies
E_reaction_theo = E_H2_theo - (2 * E_H_theo)
E_reaction_exp = E_H2_exp - (2 * E_H_exp)
# Set up the plot
fig, ax = plt.subplots(figsize=(8, 6))
ax.set_xlim(0, 3)
ax.set_ylim(-1.16, -0.93) # Adjust y-axis range to highlight differences
ax.set_xticks([])
ax.set_ylabel("Energy (Hartree)")
ax.set_title("H + H → H₂ Reaction Energy Diagram")
# Plot theoretical energy levels
ax.hlines(
y=2 * E_H_theo, xmin=0.5, xmax=1.3, linewidth=2, color="r", label="2H (Exact)"
)
ax.hlines(y=E_H2_theo, xmin=1.3, xmax=2, linewidth=2, color="b", label="H₂ (Exact)")
# Plot experimental energy levels
ax.hlines(
y=2 * E_H_exp,
xmin=0.5,
xmax=1.5,
linewidth=2,
color="r",
linestyle="dashed",
label="2H (VQE)",
)
ax.hlines(
y=E_H2_exp,
xmin=1.5,
xmax=2.5,
linewidth=2,
color="b",
linestyle="dashed",
label="H₂ (VQE)",
)
# Add labels
ax.text(
1,
2 * E_H_theo,
f"2H: {2*E_H_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
2,
E_H2_theo,
f"H₂: {E_H2_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
1,
2 * E_H_exp,
f"2H_VQE: {2*E_H_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
ax.text(
2,
E_H2_exp,
f"H₂_VQE: {E_H2_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
# Add arrows for reaction energy with ΔE label in the middle
mid_y_theo = (2 * E_H_theo + E_H2_theo) / 2
mid_y_exp = (2 * E_H_exp + E_H2_exp) / 2
ax.annotate(
"",
xy=(1.3, E_H2_theo),
xytext=(1.3, 2 * E_H_theo),
arrowprops=dict(arrowstyle="<->", color="g"),
)
ax.text(
1.35, mid_y_theo, f"ΔE: {E_reaction_theo:.4f}", color="g", verticalalignment="top"
)
ax.annotate(
"",
xy=(1.5, E_H2_exp),
xytext=(1.5, 2 * E_H_exp),
arrowprops=dict(arrowstyle="<->", color="g", linestyle="dashed"),
)
ax.text(
1.55,
mid_y_exp,
f"ΔE_VQE: {E_reaction_exp:.4f}",
color="g",
verticalalignment="center",
)
# Add legend
ax.legend()
plt.show()

Seperti yang ditunjukkan pada gambar, meskipun ada beberapa kesalahan, energi ground state eksak dari Hamiltonian dan energi reaksi yang dihitung menggunakan hasil VQE mirip, mendekati -0,2 Hartree.
Perlu dicatat di sini bahwa energi reaksi dari proses ini bernilai negatif, yang berarti energi dilepaskan melalui proses tersebut, dan molekul yang dihasilkan memiliki energi yang lebih rendah dibandingkan dua atom tunggal. 6. Kesimpulan
Mari kita rangkum apa yang sudah kita pelajari sejauh ini.
Pertama kita melihat dua teknik aproksimasi penting yang diperlukan untuk memecahkan masalah kimia kuantum: prinsip variasional dan pilihan basis set, yang keduanya merupakan dasar dari VQE. Kita menjelajahi prinsip variasional secara langsung, menghitung energi ground state dari osilator harmonik sederhana.
Selanjutnya, kita menjelajahi VQE, algoritma yang banyak digunakan untuk menghitung energi ground state dari sistem kuantum. Kita menjalankan kode untuk menghitung energi ground state untuk hidrogen atom () dan molekul hidrogen (). Khususnya, kita belajar bahwa diperlukan untuk mendapatkan Hamiltonian molekuler yang sesuai untuk sistem tersebut dan mengubahnya ke bentuk yang dapat dieksekusi pada komputer kuantum. Kita juga melihat bahwa ansatz, sebuah sirkuit kuantum terparameterisasi, diperlukan untuk menyiapkan trial quantum states dalam VQE, dan kita mendiskusikan pentingnya memilih struktur Circuit ansatz yang tepat. Kita juga belajar bahwa VQE bergantung pada proses optimisasi iteratif menggunakan komputer klasik, membimbing Circuit kuantum untuk menemukan keadaan energi terendah, dan melihat bagaimana proses tersebut konvergen.
Terakhir, kita menggunakan energi ground state yang dihitung dari dan yang diperoleh melalui VQE untuk menghitung energi reaksi pada proses .
VQE adalah algoritma kuantum near-term yang powerful, tetapi penting untuk menyadari keterbatasannya. Kinerja VQE sangat bergantung pada pilihan ansatz – menemukan ansatz yang dapat disiapkan secara efisien dan dapat merepresentasikan ground state yang sebenarnya secara akurat menjadi tantangan untuk molekul yang lebih besar dan lebih kompleks. Selain itu, perangkat keras kuantum saat ini rentan terhadap noise, yang dapat memengaruhi akurasi hasil VQE, terutama untuk Circuit yang lebih dalam atau jumlah qubit yang lebih banyak. Meskipun ada tantangan-tantangan ini, VQE berfungsi sebagai algoritma fondasi, dan penelitian yang sedang berlangsung mengeksplorasi metode variasional yang lebih canggih dan teknik mitigasi error untuk mendorong batas-batas apa yang mungkin dilakukan dalam kimia kuantum pada komputer kuantum near-term. Misalnya, algoritma seperti Sample-based Quantum Diagonalization (SQD) sedang dikembangkan, yang memanfaatkan sampel yang diperoleh dari Circuit kuantum dikombinasikan dengan diagonalisasi klasik dalam subspace untuk meningkatkan estimasi energi dan mengatasi beberapa keterbatasan yang dihadapi VQE, terutama terkait efisiensi pengukuran dan ketahanan terhadap noise.
Ulasan dan pertanyaan
Konsep penting:
- Algoritma kuantum variasional adalah paradigma komputasi di mana komputer klasik dan komputer kuantum bekerja sama untuk memecahkan suatu masalah.
- Dalam VQE, kita mulai dengan Hamiltonian dari sistem kita dan memetakannya ke qubit untuk dieksekusi pada komputer kuantum. Kita memilih Circuit kuantum terparameterisasi, sebuah ansatz, dan membuat pengukuran berulang, memvariasikan parameter ansatz, sampai nilai energi terendah tercapai. Pencarian melalui ruang parameter dilakukan menggunakan optimizer klasik. Untuk mencapai hasil yang baik, diperlukan pemilihan ansatz yang baik dan optimizer yang tepat.
- Energi reaksi adalah perubahan energi total dalam reaksi kimia, ditentukan oleh perbedaan antara energi reaktan dan produk.
Benar/salah
- Prinsip variasional menyatakan bahwa nilai ekspektasi energi untuk setiap trial wave function selalu lebih besar atau sama dengan energi ground state yang sebenarnya.
- Basis set adalah kumpulan fungsi yang digunakan untuk mengaproksimasi quantum wave functions.
- VQE adalah algoritma kuantum yang digunakan untuk menyelesaikan secara eksak persamaan Schrödinger untuk Hamiltonian tertentu.
- Dalam VQE, Circuit kuantum terparameterisasi (sebuah ansatz) digunakan untuk menyiapkan trial wave functions.
- Pilihan optimizer dalam VQE (misalnya, COBYLA, SPSA, atau ADAM) tidak memengaruhi kualitas hasil.
EstimatorQiskit digunakan untuk langsung menghitung nilai ekspektasi Hamiltonian dalam VQE.
Pertanyaan pilihan ganda:
- Apa tujuan Hamiltonian dalam VQE?
- A) Untuk menghasilkan keadaan kuantum acak
- B) Untuk menentukan energi keadaan kuantum
- C) Untuk mengoptimalkan Circuit kuantum
- D) Untuk menciptakan entanglement
- Apa tujuan utama dari algoritma VQE?
- A) Untuk menemukan energi ground state dari sebuah Hamiltonian
- B) Untuk menciptakan entanglement antar qubit
- C) Untuk melakukan pencarian Grover
- D) Untuk membobol enkripsi RSA
- Berapa banyak keadaan kuantum yang dihasilkan dalam notebook ini untuk membandingkan ansatz?
- A) 100
- B) 1000
- C) 5000
- D) 10.000
- Mengapa optimizer klasik diperlukan dalam VQE?
- A) Untuk melakukan pengukuran kuantum
- B) Memperbarui parameter ansatz untuk meminimalkan energi
- C) Untuk membuat entanglement antar qubit
- D) Untuk menghasilkan keacakan kuantum
- Mengapa ansatz dirancang untuk terparameterisasi?
- A) Untuk memungkinkan persiapan keadaan kuantum
- B) Untuk memungkinkan pencarian pada ruang keadaan kuantum yang luas
- C) Untuk mengurangi kompleksitas Circuit
- D) Untuk mengukur eigenvalue secara langsung
- Mana pernyataan yang paling benar tentang memilih ansatz yang baik?
- A) Ansatz harus menghasilkan keadaan yang terdistribusi merata di Bloch sphere, atau akan gagal.
- B) Ansatz harus disesuaikan dengan sistemmu untuk memastikan dapat menghasilkan keadaan yang mendekati ground state.
- C) Ansatz harus menghasilkan keadaan acak menggunakan parameter variasionalnya.
- D) Ansatz yang lebih baik selalu memiliki lebih banyak parameter variasional.
(Opsional) Lampiran: Overhead optimizer berdasarkan kompleksitas ansatz
VQE menghadapi beberapa tantangan yang sudah dikenal[ref 6], dan berikut ini terkait dengan apa yang sudah kita pelajari di atas.
- Tantangan pemilihan ansatz
Ada tantangan inheren dalam memilih ansatz variasional yang tepat. Ansätze yang terinspirasi kimia (seperti UCCSD) memberikan akurasi fisik tetapi membutuhkan Circuit yang dalam, sementara ansätze yang efisien untuk perangkat keras memiliki Circuit yang lebih dangkal tetapi mungkin kurang dapat diinterpretasikan secara fisik. Selain itu, banyak ansätze yang memperkenalkan parameter variasional berlebihan yang berkontribusi sedikit untuk meningkatkan akurasi tetapi secara signifikan meningkatkan kesulitan optimisasi.
- Kesulitan optimisasi
Lanskap optimisasi VQE dapat memiliki region di mana gradien menghilang secara eksponensial (barren plateaus), membuat optimizer klasik sulit untuk memperbarui parameter variasional secara efisien. Untuk ini, peneliti telah mencoba menggunakan berbagai jenis optimizer - berbasis gradien dan bebas gradien, tetapi keduanya menghadapi tantangan. Optimizer berbasis gradien menderita dari barren plateaus, sementara metode bebas gradien membutuhkan sejumlah besar evaluasi fungsi.
- Overhead optimizer
Satu lagi tantangan yang sudah dikenal adalah overhead optimizer, yang terkait dengan skala masalah. Circuit kuantum yang diperlukan untuk VQE bertumbuh dalam kedalaman dan kompleksitas seiring bertambahnya ukuran masalah; ini biasanya juga meningkatkan jumlah parameter yang perlu dioptimalkan. Proses optimisasi menjadi tidak traktabel seiring bertambahnya jumlah parameter, menyebabkan konvergensi yang lambat dan kesulitan dalam menemukan solusi optimal.
Di sini kita akan melihat tantangan-tantangan ini dengan menggunakan VQE untuk molekul , dengan dua jenis ansätze yang berbeda.
(Catatan: Ini bisa membutuhkan lebih banyak waktu QPU, jadi silakan gunakan simulator untuk ini jika kamu tidak punya cukup waktu.)
from qiskit.circuit import ParameterVector
num_iter = 4
alpha = ParameterVector("alpha", 3)
beta = ParameterVector("beta", 3 * num_iter)
# step1: Map problem to quantum circuits and operators
hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
ansatz_1 = ansatz3
ansatz_2 = QuantumCircuit(1)
for i in range(num_iter):
ansatz_2.rx(beta[i * 3 + 0], 0)
ansatz_2.rz(beta[i * 3 + 1], 0)
ansatz_2.rx(beta[i * 3 + 2], 0)
ansatz_1.draw("mpl")
ansatz_2.draw("mpl")

# Step 2: Optimize for target hardware
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa_1 = pm.run(ansatz_1)
ansatz_isa_2 = pm.run(ansatz_2)
hamiltonian_isa_1 = hamiltonian.apply_layout(layout=ansatz_isa_1.layout)
hamiltonian_isa_2 = hamiltonian.apply_layout(layout=ansatz_isa_2.layout)
Sekarang mari kita jalankan VQE dengan initial point yang terdiri dari semua satu, dengan maksimum 20 langkah, dan bandingkan konvergensi kedua run.
# QPU time 3m 40s for ibm_brisbane
# Step 3: Execute on target hardware
from scipy.optimize import minimize
x0 = np.ones(ansatz_1.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_1, hamiltonian_isa_1, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.8782202668652658]
Iters. done: 2 [Current cost: -0.43473160695469165]
Iters. done: 3 [Current cost: -0.4076372093159749]
Iters. done: 4 [Current cost: -1.3587839859772106]
Iters. done: 5 [Current cost: -1.774529906754082]
Iters. done: 6 [Current cost: -1.541934983115727]
Iters. done: 7 [Current cost: -1.2732403113465345]
Iters. done: 8 [Current cost: -1.820842221085785]
Iters. done: 9 [Current cost: -1.8065762857059005]
Iters. done: 10 [Current cost: -1.8126394095981146]
Iters. done: 11 [Current cost: -1.8205831886180421]
Iters. done: 12 [Current cost: -1.8086715778994924]
Iters. done: 13 [Current cost: -1.8307676638629322]
Iters. done: 14 [Current cost: -1.8177328827556327]
Iters. done: 15 [Current cost: -1.8179426218088064]
Iters. done: 16 [Current cost: -1.8109239667991088]
Iters. done: 17 [Current cost: -1.824271872489647]
Iters. done: 18 [Current cost: -1.813167587671394]
Iters. done: 19 [Current cost: -1.824647343397313]
Iters. done: 20 [Current cost: -1.8219785311686143]
# Save Cost_history as a new list
ansatz_1_history = cost_history_dict["cost_history"]
# QPU time 3m 40s for ibm_brisbane
x0 = np.ones(ansatz_2.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_2, hamiltonian_isa_2, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.738191173881188]
Iters. done: 2 [Current cost: -0.42636037194506304]
Iters. done: 3 [Current cost: -1.3503788613797374]
Iters. done: 4 [Current cost: -0.9109204349776897]
Iters. done: 5 [Current cost: -0.9060873157510835]
Iters. done: 6 [Current cost: -0.7735065414083984]
Iters. done: 7 [Current cost: -1.586889197437709]
Iters. done: 8 [Current cost: -1.659215191584943]
Iters. done: 9 [Current cost: -1.245445981794618]
Iters. done: 10 [Current cost: -1.1608385766138023]
Iters. done: 11 [Current cost: -1.1551733876027737]
Iters. done: 12 [Current cost: -1.8143337768286332]
Iters. done: 13 [Current cost: -1.2510951563756598]
Iters. done: 14 [Current cost: -1.6918311531865413]
Iters. done: 15 [Current cost: -1.8163783305531838]
Iters. done: 16 [Current cost: -1.8434877732947152]
Iters. done: 17 [Current cost: -1.8461898233304472]
Iters. done: 18 [Current cost: -1.0346471214915485]
Iters. done: 19 [Current cost: -1.8322518854150687]
Iters. done: 20 [Current cost: -1.717144678705999]
ansatz_2_history = cost_history_dict["cost_history"]
fig, ax = plt.subplots()
# Define the constant function)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_1_history,
label="Ansatz with 3 parameters",
)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_2_history,
label="Ansatz with 12 parameters",
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
plt.legend()
plt.draw()

Grafik di atas dengan jelas menunjukkan bahwa proses optimisasi ansatz dengan lebih banyak variabel membutuhkan lebih banyak waktu untuk mencapai konvergensi yang stabil.
Daripada mengandalkan Circuit single-qubit sederhana dan ansatz yang straightforward, kompleksitas optimisasi meningkat ketika Circuit kuantum yang lebih besar dan ansätze dengan struktur yang lebih kompleks diperlukan. Ini menyoroti tantangan yang sudah dikenal dalam VQE: overhead dari optimizer.
Para peneliti terus mengembangkan berbagai metodologi canggih yang dapat menggunakan komputer kuantum untuk masalah kimia. Kamu bisa mengakses berbagai materi pendidikan di IBM Quantum Learning.
Referensi
- [ref 1 ] Richard P. Feynman, Simulating Physics with Computers, International Journal of Theoretical Physics, 1982.
- [ref 2] Marov, M.Y. (2015). The Structure of the Universe. In: The Fundamentals of Modern Astrophysics. Springer, New York, NY.
- [ref 3] How to solve difficult chemical engineering problems with quantum computing, IBM Research Blog, 2023.
- [ref 4] Y. Cao, J. Romero and A. Aspuru-Guzik, "Potential of quantum computing for drug discovery," in IBM Journal of Research and Development, vol. 62, no. 6, pp. 6:1-6:20, 1 Nov.-Dec. 2018
- [ref 5] Present State of Molecular Structure Calculation, REv. Mod. Phys. 32, 170, 1960
- [ref 6] Fedorov, D.A., Peng, B., Govind, N. et al. VQE method: a short survey and recent developments. Mater Theory 6, 2 (2022)